Ein Leitfaden für Bindehauttumoren
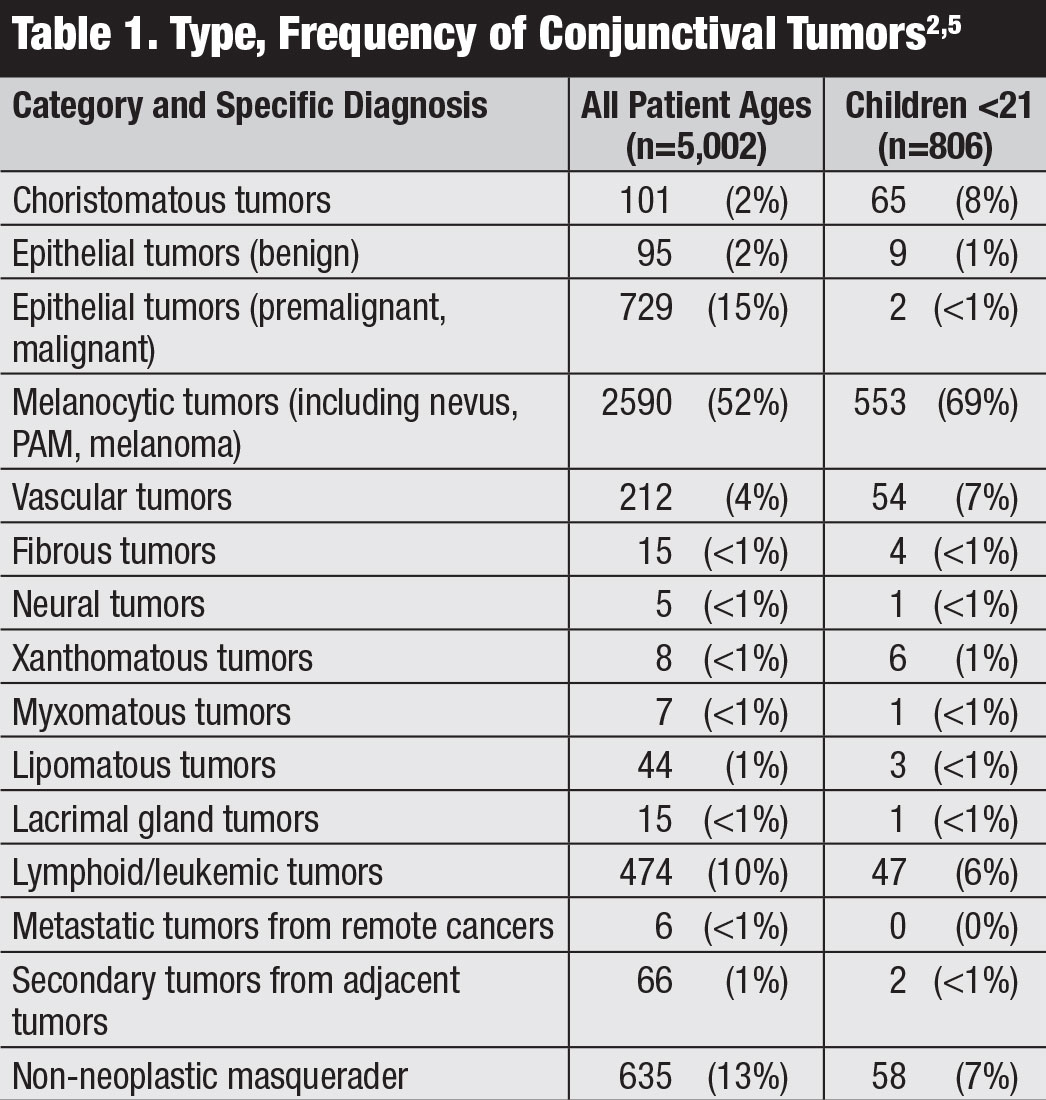
Bindehauttumoren umfassen ein Spektrum gutartiger und bösartiger Neoplasmen.1-5 Die Typen unterscheiden sich je nach Alter und Rasse, systemischem Immunstatus und Langzeitexposition. Eine große Studie mit 5.002 Fällen aus einem Augenonkologiezentrum ergab, dass 52% gutartig, 18% prämalignan und 30% bösartig waren (Tabelle 1).1,2 Obwohl dieser Bericht aus einem Zentrum für Augenonkologie stammte und maligne Erkrankungen möglicherweise überrepräsentiert sind, ist es für Kliniker wichtig, die Vielfalt der Bindehauttumoren zu verstehen.
Die fünf häufigsten Tumoren waren Nävus (23%), plattenepitheliale Neoplasie der Augenoberfläche (OSSN, 14%), primär erworbene Melanose (PAM, 12%), Melanom (12%) und lymphatischer Tumor (9%).5 Bösartige Tumoren wurden am häufigsten bei Erwachsenen beobachtet und umfassten Melanome (12%), Plattenepithelkarzinome (SCC, 9%), Lymphome (7%), Kaposi-Sarkome (<1%), Metastasen (< 1%) und andere.1 Bindehauttumoren bei Kindern zeigen nur 3% der Zeit Malignität.5
Diese Übersicht über die häufigsten Bindehauttumoren bereitet Sie darauf vor, sie angemessen zu behandeln, sei es in Ihrem Büro oder durch Überweisung.
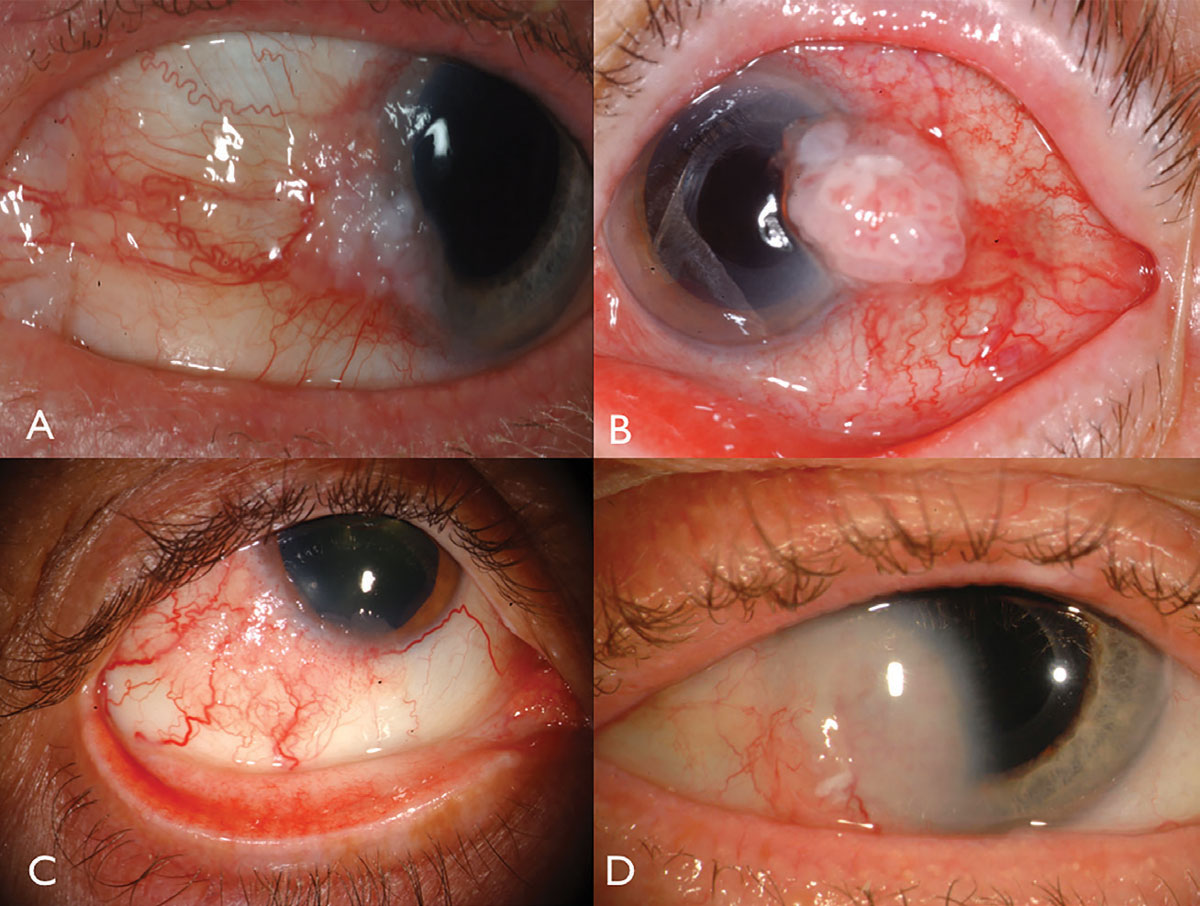
Abb. 1. Limbale OSSN: mit Leukoplakie und Hornhautbeteiligung (A), mit ausgeprägter intrinsischer Vaskularität und Feeder-Gefäßen (B) bei einem HIV-Patienten (C) und mit tiefer Hornhautinvasion, die eine Resektion und Plaque-Strahlentherapie erfordert (D). Klicken Sie auf das Bild, um es zu vergrößern.
Plattenepitheliale Neoplasie der Augenoberfläche
Der allgemeine klinische Begriff OSSN umfasst ein Spektrum von Malignomen, das von leichten epithelialen dysplastischen Veränderungen wie der intraepithelialen Neoplasie der Bindehaut (CIN) bis hin zu schwereren invasiven Karzinomen reicht, die durch die Basalmembran in die Substantia propria eindringen, wie das Plattenepithelkarzinom.
Klinische Merkmale. Konjunktivales OSSN tritt klassisch bei älteren kaukasischen Männern auf, insbesondere bei denen mit chronischer Sonnenexposition. In den Vereinigten Staaten ist Bindehaut-SCC fünfmal häufiger bei Männern und Kaukasiern. In Afrika ist Bindehaut-SCC jedoch bei Männern und Frauen fast gleich häufig und tritt in einem jüngeren Alter auf als in den Vereinigten Staaten.6
Die Plattenepithelneoplasie der Augenoberfläche tritt normalerweise als einseitige, vaskularisierte gallertartige Masse auf, die sich in der sonnenexponierten Bindehaut am nasalen oder temporalen Limbus befindet (Abbildung 1). Darüber liegende Leukoplakie, erweiterte Blutgefäße und schaumige Infiltration des angrenzenden Hornhautepithels können auftreten und selten in den Globus oder die Umlaufbahn eindringen.
Klicken Sie auf die Tabelle, um sie zu vergrößern.
Prädisponierende Faktoren. Zu den wichtigsten Umweltfaktoren für OSSN gehören chronische Sonneneinstrahlung und Zigarettenrauchbelastung. Zwei wichtige prädisponierende Faktoren des Wirts sind der helle Teint und das zugrunde liegende humane Immundefizienzvirus (HIV) und das humane Papillomavirus.6 Patienten mit Immunschwäche, insbesondere solche mit HIV, sind einem OSSN-Risiko ausgesetzt und können fortgeschrittene, bilaterale und invasive Tumoren aufweisen.1 Dies ist besonders in Afrika zu beobachten, wo HIV weit verbreitet ist und OSSN sowohl bei Männern als auch bei Frauen und in einem jüngeren Alter auftritt.6 Andere Immundysregulationen können einen Patienten für OSSN prädisponieren, einschließlich Organtransplantationsimmunsuppression, Ekzem / Atopie, Augennarbenpemphigoid, Xeroderma pigmentosum und Autoimmunerkrankungen.7
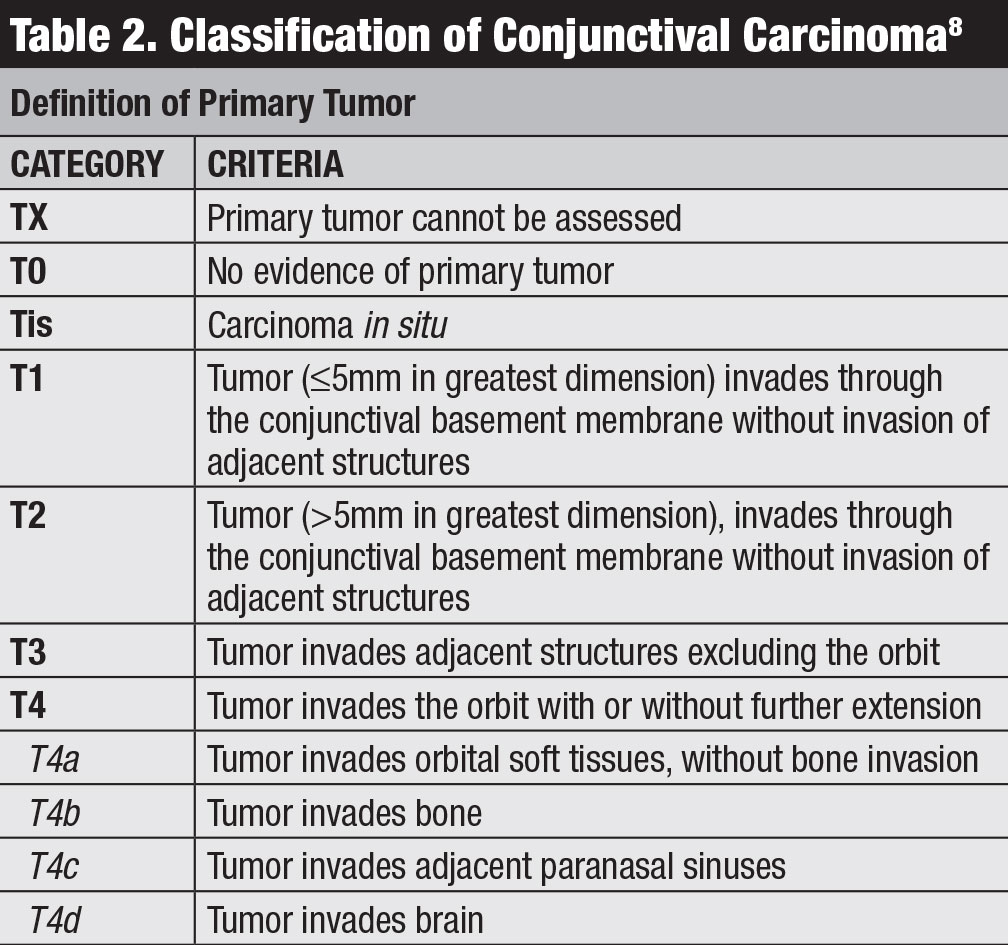
Klassifizierung. Das Handbuch des American Joint Committee on Cancer (AJCC) in der 8. Auflage enthält die neueste Klassifizierung für Bindehautkarzinome, einschließlich SCC und CIN (Tabelle 2).8
Verwaltung. Dies beinhaltet eine chirurgische Resektion mit der “No-Touch” -Technik oder nicht-chirurgische Therapien wie topische Chemotherapie mit Mitomycin C (MMC) oder 5-Fluorouracil (5-FU), topische oder injizierte Immuntherapie mit Interferon alpha-2b (IFN), topische antivirale Medikamente (Cidofovir) oder photodynamische Therapie.7,9-11
Die chirurgische No-Touch-Technik beinhaltet eine detaillierte Bewertung des Tumors mittels Spaltlampen-Biomikroskopie, um alle Tumorränder, einschließlich bulbärer, fornicealer und Tarsalkomponenten, zu visualisieren, um das gesamte Ausmaß des Tumors zu verstehen und es dem Kliniker zu ermöglichen, eine Schablonenaufnahme von Hand zu zeichnen.9 Diese Schablone wird dann in die Operation aufgenommen, um sicherzustellen, dass der gesamte Tumor entfernt wird.
Zum Zeitpunkt der Operation wird nur das umgebende normale Gewebe mit einer Pinzette gehalten, und der Tumor wird niemals berührt, um eine Aussaat des Tumors zu vermeiden. Darüber hinaus wird während der Operation keine ausgewogene Salzlösung verwendet, um eine Flüssigkeitsdispersion von Krebszellen zu vermeiden. Nach der Tumorentfernung ist der Verschluss mit sauberen Instrumenten entscheidend. Mit dieser Technik für OSSN wird Tumorpersistenz oder Rezidiv in weniger als 5% der Fälle gefunden.
Klicken Sie auf die Tabelle, um sie zu vergrößern.
Die topische Chemotherapie mit 5-FU oder MMC löst das OSSN effizient auf, häufig innerhalb von zwei bis vier Wochen nach der Therapie, obwohl ein Risiko für einen Stammzellmangel besteht. Unsere topische Therapiepräferenz ist die Immuntherapie mit IFN, da sie bei guter Tumorkontrolle gut verträglich ist, oft über drei Monate und mit geringen Komplikationen und nur geringer follikulärer Konjunktivitis.10,11 Diese Medikamente können lokal toxisch für das Hornhautepithel sein, aber weniger mit Interferon, und die Patienten sollten während der Behandlung genau beobachtet werden. Wenn die Kosten für den Patienten ein Faktor sind, ist topisches 5-FU am wenigsten teuer, gefolgt von MMC und dann IFN.
Konjunktivale lymphoide Tumoren
Lymphoide Neoplasmen reichen von niedrig- bis hochgradigen Tumoren und entstehen durch monoklonale Proliferation von Lymphozyten. Die lymphoiden Tumoren, die in der periokularen Region auftreten, betreffen häufig mehrere Gewebe wie Bindehaut, Augenhöhle und Augenlid und werden als “okuläre adnexale” lymphoide Tumoren bezeichnet, einschließlich benigner reaktiver lymphoider Hyperplasie (BRLH) und Lymphom.
BRLH und Lymphom befinden sich an entgegengesetzten Enden des Spektrums, wobei BRLH klinisch als lokalisiertes “Lachsfleck” und histopathologisch gutartig auftritt, während Lymphom auch als “Lachsfleck” auftritt, jedoch mit aggressiveren histopathologischen Merkmalen, mit mitotischer Aktivität und klassifiziert als bösartig.
Okuläre lymphatische Adnextumoren sind typischerweise B-zellulären Ursprungs. Eine multizentrische Studie mit 268 Patienten mit konjunktivalem Lymphom ergab, dass die vier häufigsten Arten das extranodale Randzonenlymphom (ENMZL, früher als schleimhautassoziiertes lymphatisches Gewebe bezeichnet) in 68%, das follikuläre Lymphom (FL) in 16%, das Mantelzell-Lymphom (MCL) in 7% und das diffuse große B-Zell-Lymphom (DLBCL) in 5%.12 Andere Arten von konjunktivalem Lymphom umfassen lymphoplasmazytisches Lymphom und Plasmazytom.
Klinische Merkmale. Das konjunktivale Lymphom tritt normalerweise bei älteren Patienten im Alter zwischen 60 und 70 Jahren auf. Dieser Tumor kann sich als primäres Lymphom manifestieren, das auf die periokulare Region beschränkt ist, oder als sekundäres Lymphom mit Krankheit an anderer Stelle. Die meisten primären Lymphome treten bei ENMZL und FL und sekundäre Lymphome bei DLBCL und MCL auf. Eine Analyse von 117 Patienten mit Bindehautlymphom ergab eine systemische Beteiligung bei 31%, am häufigsten bei Patienten mit bilateralem multifokalem okulärem Adnexlymphom.13
Das konjunktivale Lymphom manifestiert sich klassisch als rosa lachsfarbene, glatte subkonjunktivale Masse, manchmal mit Feeder-Gefäßen (Abbildung 2). Diese glatte, multilobulierte Masse kann einer follikulären oder papillären Konjunktivitis ähneln. Dieser Tumor befindet sich am häufigsten in der konjunktivalen Fornix- (44%) oder midbulbären (42%) Region und selten im Karunkel (7%) oder Limbus (7%).13 Zusätzlich zur Bindehaut kann ein Lymphom gefunden werden, das die Augenhöhle, das Augenlid oder die Uvea infiltriert.13 Die meisten Patienten mit konjunktivalem Lymphom weisen keine intraokulare Komponente auf, aber wenn vorhanden, befindet sie sich im Allgemeinen in der Uvea und nicht in der Netzhaut oder im Glaskörper.
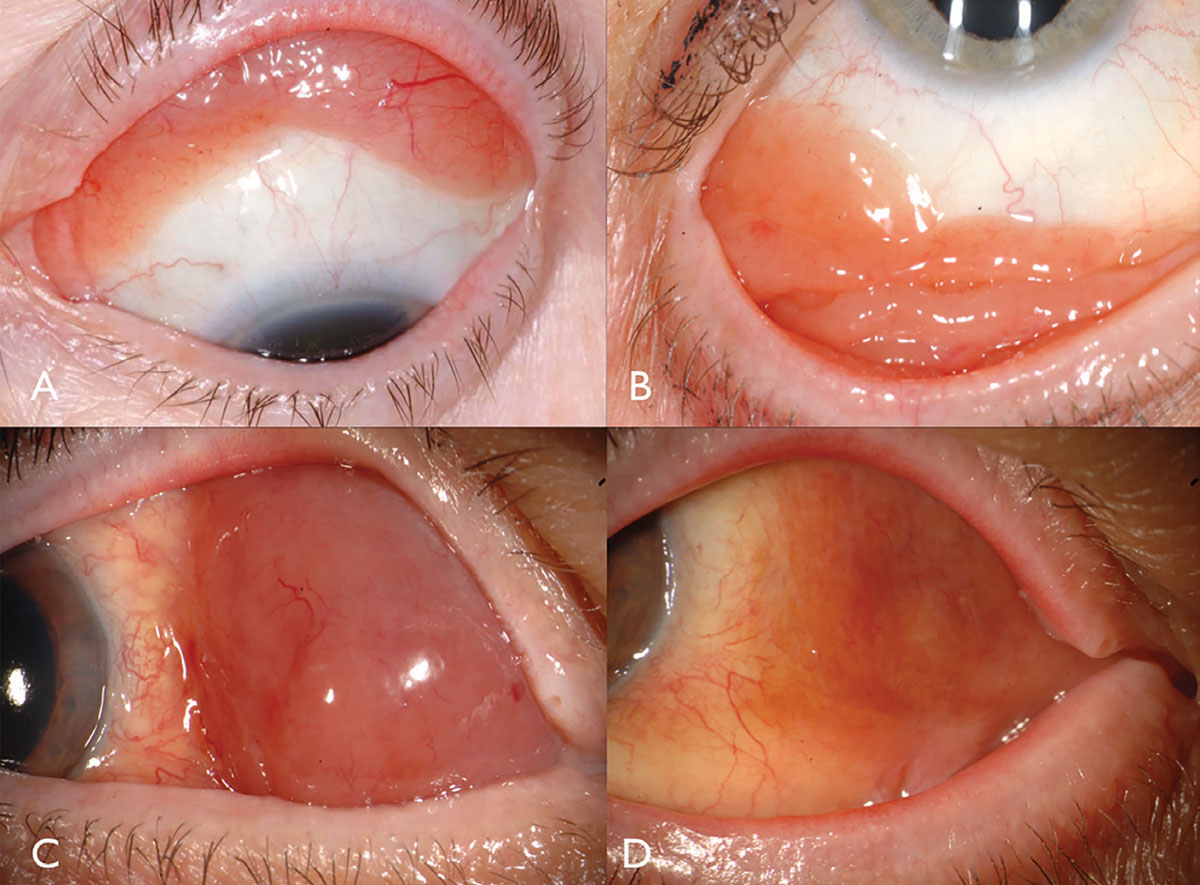
Fig. 2. Conjunctival lymphoma can be salmon-pink (A) or multilobulated forniceal (B). Medial forniceal conjunctival lymphoma before (C) and after (D) ritiximab. Click image to enlarge.
Predisposing factors. Immune dysfunction and autoimmune conditions, as well as infective etiologies such as Helicobacter pylori and Chlamydia psittaci are all predisposing factors for conjunctival lymphoma. BRLH kann ein potenzieller Vorläufer von Lymphomen sein und, während überwiegend bei Erwachsenen gefunden, kann gelegentlich bei Kindern auftreten.5 Je jünger der Patient zum Zeitpunkt der Diagnose eines lymphatischen Bindehauttumors ist, desto wahrscheinlicher ist es, dass es sich um BRLH und nicht um ein Lymphom handelt.
Klassifizierung. Es gibt mehrere Klassifikationen für konjunktivales Lymphom, einschließlich der Ann Arbor, Weltgesundheitsorganisation und AJCC 8th Edition Staging (Tabelle 3).8 Das klinische AJCC-Staging basiert auf Tumorlokalisation, regionalem Lymphknoten und entfernter Beteiligung.8
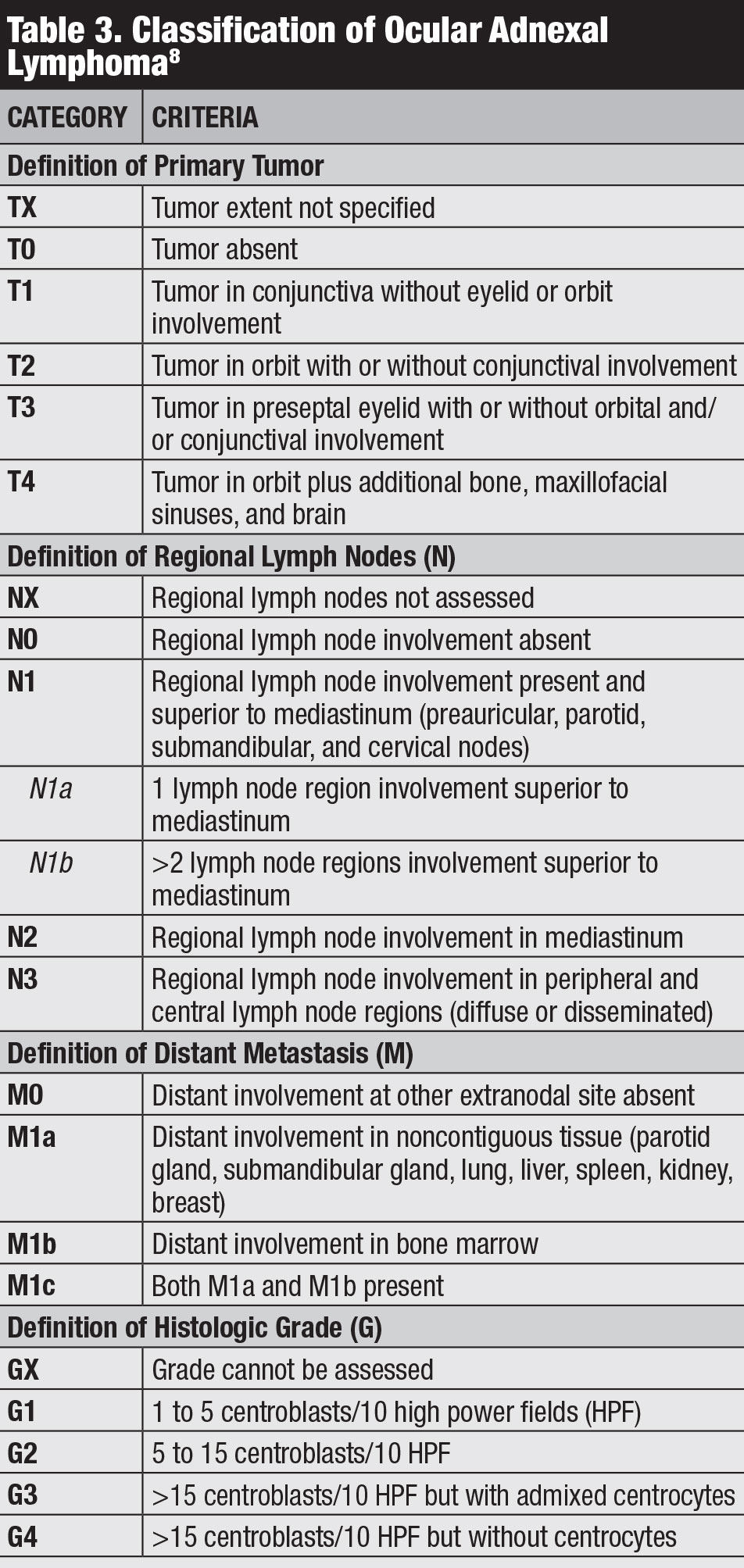
Klicken Sie auf die Tabelle, um sie zu vergrößern.
Verwaltung. Die Versorgung von Patienten mit konjunktivalem Lymphom hängt in erster Linie vom Ausmaß der periokularen Beteiligung, der systemischen Beteiligung und ihrem allgemeinen Gesundheitszustand ab. Bei Patienten mit nur konjunktivalem Lymphom und ohne systemische Beteiligung konzentriert sich die Behandlung auf eine vollständige chirurgische Resektion. Eine Behandlung mit externer Strahlentherapie oder Rituximab ist möglich, wenn der Tumor nicht resezierbar ist. Bei Patienten mit periokulärem und systemischem Lymphom ist eine Behandlung mit systemischem Rituximab oder die Zugabe einer Chemotherapie in Betracht zu ziehen.
Die systemische Prognose mit Bindehautlymphom steht in direktem Zusammenhang mit jedem Subtyp, da eine Studie ergab, dass das Fünfjahresüberleben 97% für ENMZL, 82% für FL, 55% für DLBCL und nur 9% für MCL betrug.12
Bindehautmelanom
Bindehautmelanozytentumoren sind zweifellos häufig und machen mehr als 50% der Fälle in einer großen Reihe von Bindehauttumoren aus einer Augenonkologieeinheit aus.1,2 Diese Klasse von Melanozytentumoren umfasst viele Arten wie Nävus, hautbedingte Melanose, PAM, sekundär erworbene Melanose, Melanom und Metastasen.1-5 Auf einigen Kontinenten, auf denen Patienten einen dunklen Teint haben, kann sogar OSSN melanozytisch erscheinen. Von diesen Läsionen macht der Bindehautnävus 45% und das primäre Bindehautmelanom 23% aller melanozytären Tumoren in einer augenonkologischen Praxis aus.2
In den Vereinigten Staaten verdoppelte sich die altersbereinigte Inzidenz von Bindehautmelanomen zwischen 1973 und 1999 von 0,27 pro Million auf 0,54 pro Million.14,15 Die Inzidenz stieg bei weißen Männern in den USA im gleichen Zeitraum von 27 Jahren um 295%, insbesondere bei Männern ab 60 Jahren.14 Forscher spekulieren, dass die zunehmende Rate mit der Exposition gegenüber ultraviolettem Licht zusammenhängen könnte.
Klinische Merkmale. Das Bindehautmelanom ist eine pigmentierte oder nicht pigmentierte Malignität, die durch PAM, Nävus oder de novo entstehen kann.16 Melanom kann auf der limbalen, bulbären, fornicealen oder palpebralen Bindehaut gefunden werden und zeigt oft erweiterte, gewundene und intrinsische Gefäße, die typischerweise von flachem PAM umgeben sind (Abbildung 3). Im Allgemeinen besteht bei Tumoren mit einer Dicke von mehr als 2 mm ein erhebliches Risiko für Lymphknotenmetastasen. Die Tumorinvasion in die Augenhöhle ist besonders schwerwiegend und birgt ein erhebliches Metastasierungsrisiko.
In 50% der Fälle tritt ein lokales Tumorrezidiv oder ein neuer Tumor auf, häufig im Zusammenhang mit einer neuen PAM-Transformation. Fernmetastasen – häufig in der präaurikulären, submandibulären oder zervikalen Lymphknotenkette — treten bei 25% der Patienten auf. Die Sentinel-Lymphknotenbiopsie kann Klinikern helfen, die subklinische Lymphknoteninfiltration zu bewerten. Mehrere Rezidive, insbesondere solche, die die Umlaufbahn betreffen, erfordern eine orbitale Exenteration.
Prädisponierende Faktoren. Der wichtigste prädisponierende Faktor für das Bindehautmelanom ist das Vorhandensein eines langjährigen Bindehautnävus oder PAM.16-18 Bei der Untersuchung des konjunktivalen Melanomursprungs durch Histopathologie fanden die Forscher heraus, dass der Ursprung PAM in 74%, de novo in 19% und Naevus in 7% war.16 Klinische Studien schätzen, dass sich einer von 300 Nävi zu einem Melanom entwickelt.17,18
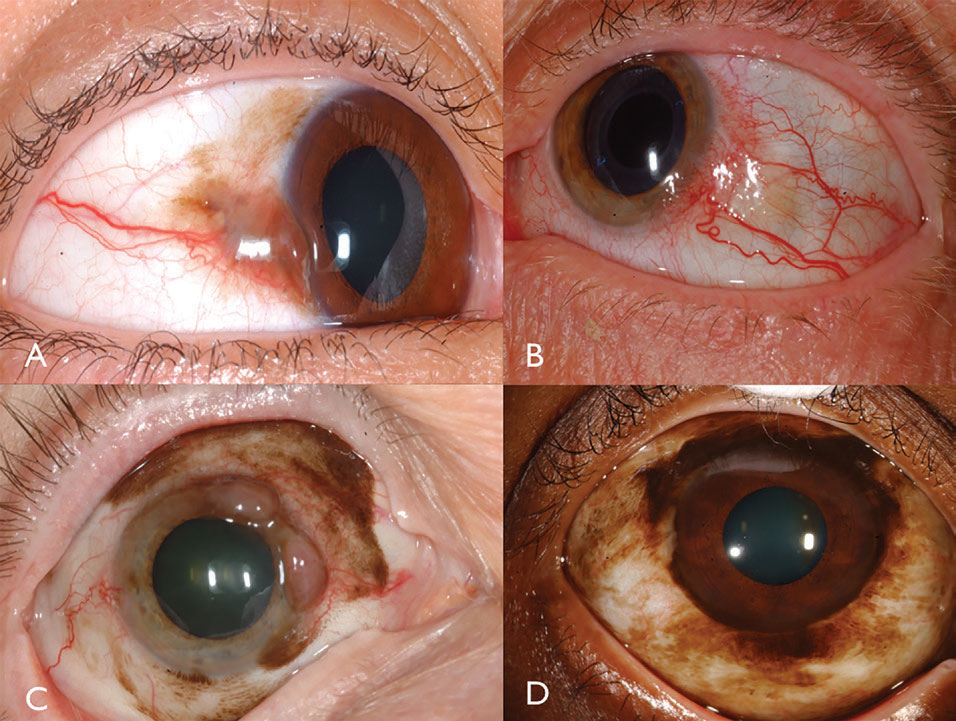
Abb. 3. Pigmentiertes Bindehautmelanom kann durch PAM (A) entstehen. Nicht pigmentiertes Bindehautmelanom kann eine intensive Vaskularität aufweisen (B). PAM kann auch ein gemischt pigmentiertes / nicht pigmentiertes Bindehautmelanom (C) verursachen. PAM verursachte bei diesem afroamerikanischen Patienten ein limbales Melanom (D). Klicken Sie auf das Bild, um es zu vergrößern.
Eine große klinische Studie ergab, dass das 10-Jahres-Risiko für die PAM-Transformation in Melanom etwa 9% betrug und das größere Ausmaß von PAM ein höheres Risiko für die Transformation in Melanom förderte.19 Daher ist es wichtig, PAM zu identifizieren und diesen Zustand mit chirurgischer Exzision, Kryotherapie und sogar oberflächlicher Keratektomie (bei Hornhautbeteiligung) zu behandeln, um Melanomen vorzubeugen.
Die Differenzierung des Bindehautnävus vom Melanom kann eine Herausforderung darstellen. In einer kürzlich durchgeführten Analyse von 510 Fällen von Bindehautnävus vs. melanom bei Kindern, Melanom war häufiger bei älteren Kindern, mit einem relativen Risiko (RR) von 4,80, größere Tumordicke (RR von 1,14), größere Basis (RR von 4,92), Tumorblutung (RR von 25,30) und fehlende intrinsische Zysten (RR von 5,06).5 Die Forscher ordneten diese Merkmale, die das Bindehautmelanom bei Kindern vorhersagen, einer Mnemonik zu: Melanom fangen, Repräsentieren: Kinderalter älter, Dicke / Basis größer, Zyste fehlt, Blutung für Melanom.5
Die Differenzierung von PAM vom Melanom kann ebenfalls schwierig sein; Melanom hat jedoch Dicke und PAM ist völlig flach. In einer Analyse von 1.224 Fällen von PAM vs. Melanom in allen Altersgruppen, Melanom mit signifikant größer basierend auf medianem Patientenalter (54 vs. 61 Jahre); männliches Geschlecht (35% vs. 49%); Lage in Fornix (2% vs. 6%) und Tarsus (1% vs. 4%); größerer medianer basaler Durchmesser (6mm vs. 8mm), Dicke (< 1mm vs. 1mm), Feeder-Gefäße (10% vs. 48%) und intrinsische Gefäße (4% vs. 33%); und Blutung (<1% vs. 3%).2
Gewebebiomarker sind wichtig für die Beurteilung des konjunktivalen Melanoms und umfassen BRAF-Mutation, TERT-Promotor-Mutation und PTEN-Mutation.1 Die Identifizierung dieser Biomarker ist bei der Planung einer systemischen Therapie zur Behandlung oder Prävention von Metastasen von entscheidender Bedeutung, da gezielte Therapien gegen bestimmte Biomarker wie Vemurafenib für BRAF-mutierte Malignome verfügbar sind.
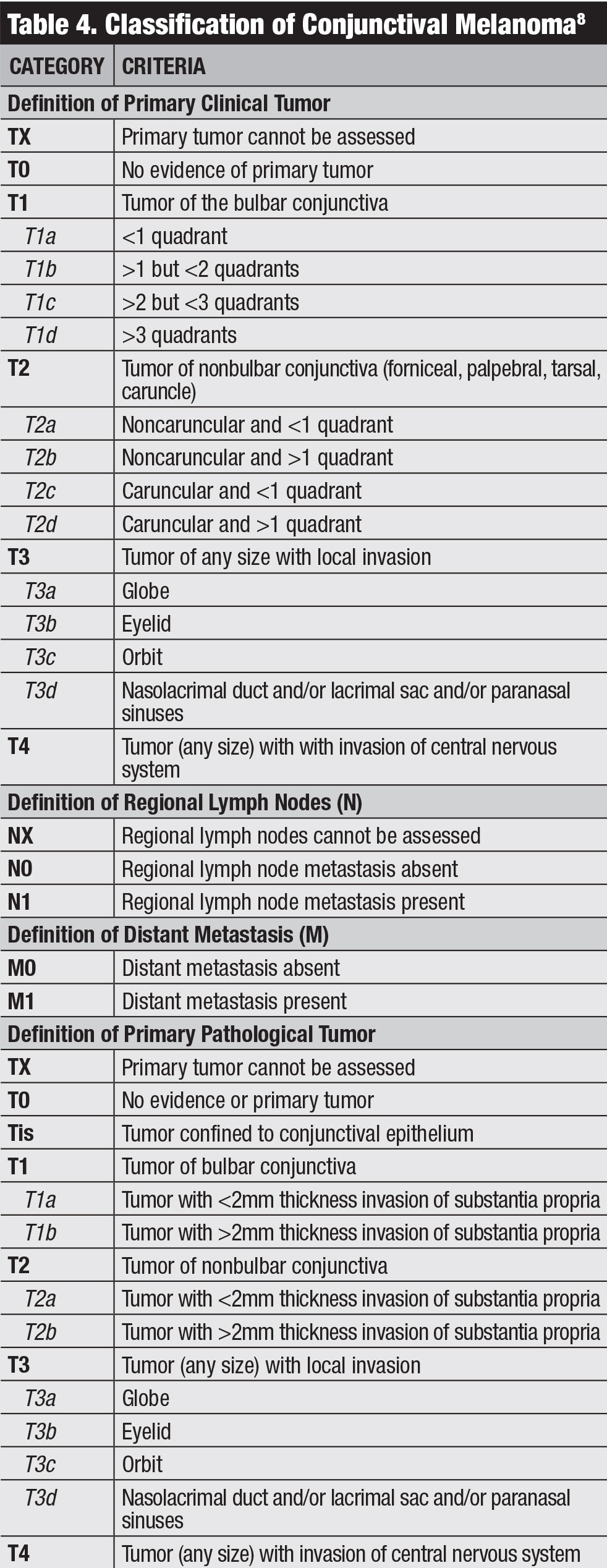
Klicken Sie auf die Tabelle, um sie zu vergrößern.
Klassifizierung. Die klinische Klassifikation des AJCC für das Bindehautmelanom basiert auf der Tumorausdehnung nach Quadranten, Tumorlokalisation und invasiven Merkmalen (Tabelle 4).8 Unser Team untersuchte die Ergebnisse des konjunktivalen Melanoms basierend auf der AJCC 7th Edition und fand heraus, dass dieses Stadium die Prognose sehr vorhersagbar war.20 Melanome, die als T2 und T3 klassifiziert wurden (im Vergleich zu T1), zeigten signifikant höhere Lokalrezidivraten, regionale Lymphknotenmetastasen, Fernmetastasen und Tod.
Verwaltung. Die Behandlung des konjunktivalen Melanoms umfasst im Wesentlichen eine vollständige chirurgische Resektion unter Verwendung der No-Touch-Technik, um eine Tumorsaat zu vermeiden. Die erste Operation ist die wichtigste, da die empfindliche Entfernung des gesamten Tumors ohne Tumorsaat der Schlüssel zur Verhinderung zukünftiger Rezidive und Metastasen ist.16
Das Melanom am Limbus corneoskleralis wird unter dem Operationsmikroskop ebenfalls mit der No-Touch-Technik entfernt. Die flache Hornhautkomponente wird mit absolutem Alkohol entfernt, oberflächliche Epitheliektomie ohne Unterbrechung der Bowman-Membran. Der Bindehautabschnitt wird mit 2 mm bis 3 mm Rändern entfernt und am Limbus durch flache episklerale Dissektion freigesetzt. Wenn eine sklerale Invasion vorliegt, wird eine Plaque-Strahlentherapie angewendet. Alle Bindehautränder werden mit doppelter Frost-Tau-Kryotherapie behandelt.
Die Rekonstruktion umfasst primäre Verschlusstechniken, Rotationslappen oder Amnionmembrantransplantation, häufig mit Symblepharonring mit Amnionmembran Drapierung. Melanome, die sich in die Umlaufbahn erstrecken, erfordern eine orbitale Exenteration oder in jüngerer Zeit eine Immuntherapie mit Checkpoint-Hemmung.21
Patienten mit Bindehautmelanom sollten von einem Augenonkologen auf Lokalrezidive und von einem systemischen Melanomonkologen auf Metastasen überwacht werden, insbesondere mit regionaler Lymphknotenpalpation und Sentinel-Lymphknotenbiopsie. Metastasen treten zunächst in den präaurikulären oder submandibulären Lymphknoten auf, später in Lunge und Gehirn. Neue Hinweise deuten darauf hin, dass Melanommetastasen empfindlich auf BRAF-Inhibitoren oder Immun-Checkpoint-Inhibitoren reagieren könnten.21,22
Bindehauttumoren umfassen ein breites Spektrum von Tumoren. Die häufigsten Malignome sind OSSN, Lymphom und Melanom. Das Erkennen der klassischen klinischen Merkmale, das Verständnis von Vorläufern und das schnelle und angemessene Management dieser Malignome sind wichtig für die besten Patientenergebnisse.
Dr. Shields, Lally und Shields arbeiten im okulären Onkologiedienst am Wills Eye Hospital der Thomas Jefferson University in Philadelphia. Unterstützt von der Eye Tumor Research Foundation, Philadelphia.
1. Shields CL, Chien JL, Surakiatchanukul T, et al. Bindehaut-Tumoren: überprüfung der klinischen Merkmale, Risiken, Biomarker und Ergebnisse. Die 2017 J. Donald M. Gass Lecture. Asien Pac J Ophthalmol. 2017;6:109-20.
2. Shields CL, Alset AE, Boal NS, et al. Bindehauttumoren in 5002 Fällen. Vergleichende Analyse von gutartigen gegenüber bösartigen Gegenstücken. Die James D. Allen Lecture 2016. In: Am J Ophthalmol. 2017;173:106-33.
3. Schilde CL, Schilde JA. Tumoren der Bindehaut und Hornhaut. In: Surv Ophthalmol. 2004;49:3-24.
4. Großniklaus HE, Grün WR, Luckenbach M, et al. Bindehautläsionen bei Erwachsenen. Eine klinische und histopathologische Überprüfung. Hornhaut. 1987;6:78-116.
5. Dr.-Ing. Dr.-Ing. Dr.-Ing. Sioufi K, Alset AE, et al. Bindehauttumoren bei Kindern. Merkmale, die gutartige von bösartigen Tumoren unterscheiden. In: JAMA Ophthalmol. 2017;135:215-24.
6. Gichuhi S, Sagoo MS, Weiss HA, et al. Epidemiologie der plattenepithelialen Neoplasie der Augenoberfläche in Afrika. In: Trop Med Int Health. 2013;18:1424-43.
7. Schilde CL, Ramasubramanian A, Mellen P, Schilde JA. Konjunktivales Plattenepithelkarzinom, das bei immunsupprimierten Patienten auftritt (Organtransplantation, Infektion mit dem humanen Immundefizienzvirus). Augenheilkunde. 2011;118:2133-7.
8. Amin MB, Rand S, Greene F, et al., eds. In: AJCC Cancer Staging Manual. 8. Aufl. Chicago; Springer International Publishing: 2017.
9. Schilde JA, Schilde CL, De, PV. Chirurgischer Ansatz bei Bindehauttumoren. Die Lynn B. McMahan Lecture von 1994. Arch Ophthalmol. 1997;115:808-15.
10. Shields CL, Kaliki S, Kim HJ, et al. Interferon für plattenepitheliale Neoplasien der Augenoberfläche in 81 Fällen: Ergebnisse basierend auf dem American Joint Committee on Cancer Classification. Hornhaut. 2013;32(3):248-56.
11. Karp CL, Galor A, Chhabra S, et al. Subkonjunktivales / periläsionales rekombinantes Interferon a2b bei plattenepithelialer Neoplasie der Augenoberfläche: eine 10-Jahres-Übersicht. Augenheilkunde. 2010;117(12):2241-6.
12. Kirkegaard MM, Rasmussen PK, Coupland SE, et al. Konjunktivales Lymphom – Eine internationale multizentrische retrospektive Studie. In: JAMA Ophthalmol. 2016;134:406-14.
13. Schilde CL, Schilde JA, Carvalho C, et al. Konjunktivale lymphoide Tumoren: klinische Analyse von 117 Fällen und Beziehung zum systemischen Lymphom. Augenheilkunde. 2001;108:979-84.
14. Yu GP, Hu DN, McCormick S, Finger PT. Konjunktivales Melanom: Steigt sie in den USA? In: Am J Ophthalmol. 2003;135:800-6.
15. Tuomaala S, Kivela T. Korrespondenz in Bezug auf konjunktivales Melanom: Nimmt es in den Vereinigten Staaten zu? In: Am J Ophthalmol. 2003;136:1189-90.
16. Schilde CL, Markowitz JS, Belinsky I, et al. Konjunktivales Melanom. Ergebnisse basierend auf Tumorursprung in 382 aufeinanderfolgenden Fällen. Augenheilkunde. 2011;118:389-95.
17. Shields CL, Fasiuddin AF, Mashayekhi A, et al. Konjunktivale Nävi: klinische Merkmale und natürlicher Verlauf bei 410 aufeinanderfolgenden Patienten. Arch Ophthalmol. 2004;122:167-75.
18. Gerner N, Norregaard JC, Jensen OA, Prause JU. Bindehaut-Naevi in Dänemark 1960-1980. Eine 21-jährige Follow-up-Studie. In: Acta Ophthalmol Scand. 1996;74:334-7.
19. Schilde JA, Schilde CL, Mashayekhi A, et al. Primär erworbene Melanose der Bindehaut: Risiken für das Fortschreiten zum Melanom bei 311 Augen. Die Lorenz E. Zimmerman Lecture 2006. Augenheilkunde. 2008;115:511-9.
20. Shields CL, Kaliki S, Al-Dahmash S, et al. Die klinische Klassifikation des American Joint Committee on Cancer (AJCC) sagt die Ergebnisse des konjunktivalen Melanoms voraus. Ophthalm Plast Reconstr Surg. 2012;5:313-23.
21. Kandl TJ, Thakar SD, Sagiv O, et al. Immuntherapie mit programmiertem Zelltod 1 Inhibitoren für 5 Patienten mit konjunktivalem Melanom. In: JAMA Ophthalmol. 2018 November 1;136(11): 1236-41.
22. Dalvin LA, Schilde CL, Orloff M, et al. Checkpoint-Inhibitor-Immuntherapie. Systemische Indikationen und ophthalmische Nebenwirkungen. Netzhaut. 2018;6:1063-78.