Fronteras en Neurología
Introducción
La neuroacantocitosis es un término superior para un grupo de síndromes raros que se caracterizan por síntomas neurológicos en combinación con glóbulos rojos deformados puntiagudos (acantocitos). Este informe se centra en la corea-acantocitosis (ChAc) , que es una enfermedad huérfana con aproximadamente 1.000 individuos afectados en todo el mundo causada por mutaciones autosómicas recesivas en el gen VPS13A (homólogo de clasificación de proteínas vacuolares 13) en el cromosoma 9q (1). La enfermedad tiene un curso progresivo, las causas del aumento de la mortalidad pueden ser funciones motoras disminuidas que se correlacionan con condiciones de riesgo como disfagia, pero también se han descrito muertes repentinas e inesperadas (2, 3). Hasta ahora, no hay tratamiento causal.
La coexistencia sospechosa de acantocitosis y trastornos del movimiento fue reportada por primera vez en la década de 1970 por Levine y Critchley (4, 5) y desde entonces ha sido aceptada como la característica prominente de la enfermedad. Sin embargo, en nuestro informe de caso describimos un fenotipo clínico raro de ChAc que carece de trastornos del movimiento evidentes, lo que sugiere una variedad más amplia de presentaciones clínicas. Las herramientas de diagnóstico tienen que cumplir con estos desafíos para establecer el diagnóstico correcto, aquí sugerimos la neuroimagen molecular como un método clave.
Reporte de un caso
El paciente índice masculino presentó por primera vez a los 25 años de edad dos crisis tónico-clónicas bilaterales no provocadas. No había antecedentes médicos. La resonancia magnética cerebral y el EEG a esa edad eran normales y no se inició ningún tratamiento debido a la reticencia del paciente y a eventos poco frecuentes. Sin embargo, el paciente tenía convulsiones continuas que ahora se presentaban como epilepsia parcial. Describió sensaciones recurrentes de mareos repentinos que fueron diagnosticados como un aura vertiginosa. Además, tuvo convulsiones discognitivas en las que mostró (a) falta de respuesta y recitación de oraciones turcas, (b) automatismos orales, o (c) juguetear con las manos y pronunciar sonidos, todo en parte evolucionando a convulsiones tónicas clónicas. El video-EEG ahora mostraba complejos locales de ondas de pico tanto en el lóbulo temporal derecho como izquierdo. De acuerdo con la semiología de la convulsión, se realizó el diagnóstico de epilepsia del lóbulo temporal bilateral y se inició la medicación con levetiracetam.
A la edad de 31 años, las convulsiones no se suprimieron por completo, como parte de los diagnósticos adicionales de epilepsia del lóbulo temporal, el paciente se sometió a una TEP-FDG (Figura 1) que mostró un hipometabolismo mesiotemporal bilateral, en línea con el origen de la convulsión mesiotemporal. Sin embargo, también hubo un hipometabolismo estriado marcado y muy inusual que levantó la sospecha de un trastorno neurodegenerativo del movimiento. En consecuencia, se inició una amplia evaluación de seguimiento (véase el gráfico 2). Se realizó un FP-CIT-SPECT que produjo una pérdida moderadamente bilateral de la disponibilidad del transportador de dopamina estriado, en línea con una disminución de la integridad nigroestriatal (Figura 1). Una resonancia magnética de alta resolución presentó ahora atrofia bilateral del núcleo caudado (Figura 3). El examen neurológico mostró un patrón de marcha limitado discreto e hipomimia, pero sin otra afección del sistema motor extrapiramidal y sin síntomas bulbar como distonía de alimentación. Aparte de un estado reflejo bajo en los miembros inferiores, para el cual los estudios de conducción nerviosa confirmaron una polineuropatía axonal sensomotora, el examen neurológico fue normal. Los síntomas neuropsicológicos incluyeron aspectos de un trastorno de ansiedad y el paciente informó dificultades con pérdida de memoria a corto plazo. Sin embargo, las pruebas neuropsicológicas no confirmaron un mal funcionamiento mnésico manifiesto, sino una distracción inespecífica, que, además, podría haberse visto reforzada por una supresión insuficiente de las convulsiones en ese momento. Las pruebas de laboratorio fueron negativas para cualquier epilepsia sintomática (p. ej., anticuerpos de encefalitis autoinmune, anticuerpos antineuronales). El líquido cefalorraquídeo no mostró signos de inflamación, pero sí un aumento moderado de proteínas (765 mg/l). El análisis de aminas biogénicas en el líquido cefalorraquídeo reveló una elevación de glutamina que asociamos con convulsiones recientes. En sangre periférica se encontró el 0,4% de los acantocitos. Las pruebas serológicas para la enfermedad de Wilson fueron negativas. Se notó una elevación persistente de la creatina cinasa (rango: 1,000–3,000 U/l) que llevó al diagnóstico sospechoso de un trastorno mitocondrial. Para investigaciones posteriores, se realizó una prueba de lactato isquémico que mostró resultados normales. Se realizó biopsia muscular de M. gastrocnemicus, pero el análisis de la función mitocondrial y de la cadena respiratoria fue normal.FIGURA
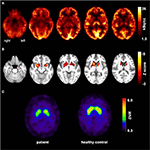
Gráfico 1 Resultados de estudios de imagen molecular. Las imágenes transaxiales de FDG-PET (A) mostraron no solo un hipometabolismo mesiotemporal bilateral leve (en línea con el origen de la convulsión mesiotemporal), sino también un marcado hipometabolismo estriado que resultó ser altamente significativo en comparación con los controles sanos . Un examen adicional de FP-CIT-SPECT (C) también reveló una pérdida moderada de transportadores de dopamina estriados, siendo testigo de una disminución de la integridad nigroestriatal (se muestra un control saludable adaptado a la edad para comparar; DVR, relación de volumen de distribución).

Figura 2. Evolución clínica y diagnóstica del paciente índice.FIGURA
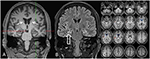
Gráfico 3 La resonancia magnética muestra atrofia de cabeza caudal bilateral discreta y un hipocampo derecho más pequeño e hiperintenso que indica esclerosis del hipocampo derecho . El hipocampo izquierdo es algo hiperintenso pero no atrófico. La atrofia de cabeza caudada bilateral se confirma con el análisis de RVC, en el que los colores azules muestran una disminución de los colores grises y rojos, un aumento del volumen de líquido cefalorraquídeo, respectivamente (C).
Los antecedentes familiares revelaron una consanguinidad de sus padres (primos hermanos), que a su vez no tenían antecedentes médicos significativos. De sus seis hermanos, dos (de entre 20 y 30 años de edad) ya habían sufrido convulsiones generales (véase la Figura 4). Los registros médicos de sus hermanos no estaban disponibles para nosotros.FIGURA
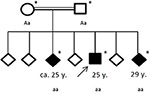
Gráfico 4 Genealogía de la familia afectada, con la edad de la primera crisis epiléptica en años (y). Flecha: patente de índice. Asterisco: pruebas genéticas realizadas. Aa, heterocigoto; aa, homocigoto para c. 4326 T> A (p. Tyr1442*).
Bajo la hipótesis de un trastorno de movimiento neurodegenerativo hereditario, el paciente y su familia fueron derivados para pruebas genéticas. La secuenciación del exoma reveló una mutación truncante homocigótica novedosa c. 4326 T> A (p. Tyr1442*) en el gen ChAc VPS13A en los tres hermanos afectados. Los padres consanguíneos se detectaron heterocigotos. No se identificaron otras variantes o mutaciones en la secuenciación del exoma.
De hecho, en un seguimiento a la edad de 33 años, el paciente índice todavía tenía síntomas extrapiramidales marginales y ninguno estaba presente en ninguna de sus hermanas. Ahora daba la impresión de una ligera rigidez solo bajo cebado en el miembro superior derecho, y una leve bradicinesia de los miembros superiores. En tratamiento con levetiracetam y lacosamida, el paciente no ha sufrido convulsiones.
Discusión
Con el presente caso, demostramos que ChAc puede manifestarse clínicamente con epilepsia del lóbulo temporal sin síntomas motores significativos debido a una nueva mutación correspondiente en el gen VPS13A.
El curso de la enfermedad se caracteriza típicamente por un trastorno progresivo del movimiento (incluyendo corea, distonía, parkinsonismo), cambios cognitivos y psiquiátricos y síntomas miopáticos con marcadores serológicos de acantocitosis e hipercemia. Las convulsiones se han observado previamente como un síntoma de ChAc (6), pero los trastornos del movimiento todavía se consideran el síntoma clave. La evidencia surge de que la epilepsia, y más específicamente la epilepsia que se origina en el lóbulo temporal, podría ser un fenotipo subestimado de ChAc. Peluso et al. reportó un paciente similar al nuestro, que, incluso a la edad de 46 años, no mostró trastornos del movimiento mientras presentaba hallazgos de neuroimagen indicativos de compromiso de ganglios basales (7). De acuerdo con eso, Scheid et al. se describieron tres pacientes con epilepsia del lóbulo temporal mesial y esclerosis (según estudios de RM) como el síntoma predominante y se les diagnosticó ChAc (8). Además, Mente et al. recientemente se proporcionaron los resultados de la autopsia de un paciente de ChAc que no solo presentaba atrofia de los ganglios basales, sino también esclerosis hipocampal (9). En línea con nuestro caso, la implicación bilateral del hipocampo parece ser un hallazgo común (10, 11). Sin embargo, la relación exacta entre las convulsiones y la esclerosis sigue siendo una cuestión de gallina o huevo. El papel correlativo de la coreína en el desarrollo estructural del hipocampo aún no se entiende completamente. Curiosamente, en un modelo de ratón de ChAc se observó un cambio estructural, ya que la expresión de la proteína hipocampal del receptor GABA (A) gamma 2 y su proteína de anclaje coreína se incrementaron (12).
En la rutina clínica, este fenotipo todavía puede ser un desafío para encontrar el diagnóstico correcto. El primer indicio crucial hacia una enfermedad neurodegenerativa en nuestra paciente fue una disminución prominente en el metabolismo de la glucosa estriada y una disminución moderada en la integridad nigroestriatal en FDG-PET y FP-CIT-SPECT, respectivamente. La pequeña colección de resultados de imagen funcional, basada en la descripción de series de casos, muestra que las alteraciones en el metabolismo y la disfunción dopaminérgica ocurren principalmente en el núcleo caudado y el putamen (13). La resonancia magnética cerebral reveló atrofia del núcleo caudado durante el curso de la enfermedad, lo que se ha descrito como un hallazgo típico en ChAc (14). En consecuencia, las características neurológicas distintivas generalmente incluyen corea, distonía de alimentación, discinesias orofaciolinguales y tics, junto con otros síntomas de afección de los ganglios basales, como parkinsonismo y distonía (3). Es tentador especular que los cambios estriados observados en nuestro paciente todavía estaban por debajo del umbral de los síntomas causantes (es decir, hallazgos de imágenes prodrómicas), que finalmente pueden ocurrir a medida que la enfermedad progresa. Por lo tanto, fomentamos las imágenes estructurales y moleculares como una herramienta de diagnóstico sensible en esta enfermedad huérfana.
En ChAc, hay descripciones de recuentos de acantocitos entre el 5 y el 50% (3), pero hay algunos informes de casos que demuestran que los acantocitos pueden aparecer solo tarde en el curso de la enfermedad (15), pueden ser muy bajos o incluso estar completamente ausentes (16). Por lo tanto, los acantocitos no deben considerarse obligatorios para hacer el diagnóstico. Además, la epilepsia podría retrasar el diagnóstico, ya que disimula otros síntomas típicos de ChAc. En particular, los síntomas psiquiátricos, como los cambios de personalidad y el deterioro cognitivo, que son muy frecuentes, pueden malinterpretarse y atribuirse a efectos secundarios relacionados con el fármaco (es decir, con levetiracetam) o a convulsiones. Se observa hipercemia secundaria después de convulsiones tónicas clónicas generales y se puede pasar por alto una elevación persistente.
Nuestro caso también destaca el beneficio crucial de la secuenciación del exoma para establecer un diagnóstico correcto, especialmente en enfermedades raras o cuando los síntomas son vagos. La familia de genes VPS13 (A–D) de mamíferos es de creciente interés y se han identificado mutaciones en otras enfermedades neurológicas, como la enfermedad de Parkinson autosómica recesiva o la ataxia espinocerebelosa autosómica recesiva (17). Las mutaciones recesivas en VPS13D causan trastornos del movimiento de inicio en la infancia (18). La fisiopatología subyacente de ChAc aún no está totalmente descifrada, pero se demostró que el VPS13A codifica la proteína coreína que se expresa de forma ubicua en el cerebro y en un gran número de otros tejidos (19). Los estudios revelan que participa en las vías de señalización que regulan la arquitectura citoesquelética, la exocitosis y la supervivencia celular (20). Otra mutación que se ha descrito que se presenta con un fenotipo dominado por convulsiones en 9 pacientes con ChAc es la mutación c. 2343del en el gen VPS13A (21). Es razonable suponer que mutaciones específicas en el mismo gen resultan en una cierta aberración de la coreína que causa un fenotipo único, por ejemplo, al afectar diferentes áreas del cerebro. Sin embargo, la fisiopatología subyacente es todavía solo especulativa y la documentación adicional de las mutaciones causantes será de interés. Mostramos aquí que la mutación truncante c. 4326 T> A (p. Tyr1442*), que no ha sido descrita anteriormente, da como resultado un fenotipo similar con epilepsia predominante en todos los miembros de la familia afectados.
Observaciones finales
Es probable que la epilepsia (especialmente la epilepsia del lóbulo temporal bilateral) sea un fenotipo predominante de ChAc. Por lo tanto, se debe realizar cuidadosamente una búsqueda de señales de alerta adicionales (como hipercemia, antecedentes familiares, polineuropatía). En casos sospechosos, se debe agregar la búsqueda de acantocitos en la sangre y el cerebro, ya que estas herramientas están ampliamente disponibles. En caso de duda, el diagnóstico debe complementarse con FDG-PET y / o FP-CIT-SPECT, ya que son sensibles a la detección de compromiso estriado. En epilepsias autosómicas recesivas con hallazgos indicativos de una enfermedad neurodegenerativa, se debe utilizar la prueba de un solo gen VPS13A o la transferencia de sangre occidental de coreína para establecer el diagnóstico correcto. Esto último también debe considerarse en otros trastornos neurodegenerativos sin etiología definida genéticamente.
Declaración ética
Se obtuvo el consentimiento informado por escrito del paciente para la participación en el estudio. Se obtuvo el consentimiento informado por escrito del paciente para la publicación de este reporte de caso.
Contribuciones de los autores
JW, RM y SK participaron en el manejo de pacientes. JW escribió el primer borrador del manuscrito. LF, HU y PM prepararon figuras. Todos los autores contribuyeron a la revisión del manuscrito, leyeron y aprobaron la versión presentada.
Declaración de conflicto de intereses
HU es accionista de Veobrain Gmbh, una empresa derivada del Centro Médico Universitario de Friburgo.
Los demás autores declaran que la investigación se realizó en ausencia de relaciones comerciales o financieras que pudieran interpretarse como un posible conflicto de intereses.
1. Walterfang M, Evans A, Leong Looi JC, Jung HH, Danek A, Walker RH, et al. La neuropsiquiatría de los síndromes de neuroacantocitosis. Neurosci Biobehav Rev. (2011) 35:1275-83. doi: 10.1016 / j. neubiorev.2011.01.001
Resumen de PubMed / Texto Completo de CrossRef / Google Scholar
2. Walker RH. Manejo de síndromes de neuroacantocitosis. Temblor Otra Hiperquiné. Mov. (2015) 5:346. doi: 10.7916 / D8W66K48
Resumen de PubMed / Texto completo de CrossRef / Google Scholar
3. Velayos Baeza A, Dobson-Stone C, Rampoldi L, Bader B, Walker RH, Danek A, et al. Corea-Acantocitosis. GeneReviews®. Seattle, WA: Universidad de Washington (1993).
4. Critchley EM, Clark DB, Wikler A. Acantocitosis y Trastorno Neurológico sin Betalipoproteinemia. Arch Neurol. (1968) 18:134–40.
Resumen de PubMed
5. Levine IM, Estes JW, Looney JM. Enfermedad neurológica hereditaria con acantocitosis. Un nuevo síndrome. Arch Neurol. (1968) 19:403–9.
Resumen de PubMed | Google Scholar
6. Hardie RJ, Pullon HW, Harding AE, Owen JS, Pires M, Daniels GL, et al. Neuroacantocitosis. estudio clínico, hematológico y patológico de 19 casos. Brain (1991) 114 (Pt 1A): 13-49.
Resumen de PubMed | Google Scholar
7. Peluso S, Bilo L, Esposito M, Antena A, Rosa ADR, Pappata S, et al. Corea-acantocitosis sin corea: expansión del fenotipo clínico. Parkinson Relat Disord. (2017) 41:124–6. doi: 10.2016 / j.parkreldis.2017.05.013
Resumen de PubMed / Texto Completo de CrossRef / Google Scholar
8. Scheid R, Bader B, Ott DV, Merkenschlager A, Danek A. Desarrollo de epilepsia del lóbulo temporal mesial en corea-acantocitosis. Neurology (2009) 73: 1419-22. doi: 10.1212 / WNL.0b013e3181bd80d4
Resumen de PubMed / Texto completo de CrossRef / Google Scholar
9. Mente K, Kim SA, Grunseich C, Hefti MM, Crary JF, Danek A, et al. Esclerosis hipocampal y epilepsia del lóbulo temporal mesial en corea-acantocitosis: un caso con evaluación clínica, patológica y genética. Neuropatol Apl Neurobiol. (2017) 43:542–6. doi: 10.1111 / nan.12403
Resumen de PubMed | Texto Completo de CrossRef / Google Scholar
10. Bader B, Vollmar C, Ackl N, Ebert A, la Fougère C, Noachtar S, et al. Epilepsia bilateral del lóbulo temporal confirmada con EEG intracraneal en corea-acantocitosis. Seizure (2011) 20:340-2. doi: 10.1016 / j.incautación.2010.12.007
Resumen de PubMed / Texto Completo de CrossRef / Google Scholar
11. Marson AM, Bucciantini E, Gentile E, Geda C. Neuroacantocitosis: hallazgos clínicos, radiológicos y neurofisiológicos en una familia italiana. Neurol Sci. (2003) 24:188–9. doi: 10.1007 / s10072-003-0123-1
Resumen de PubMed / Texto Completo de CrossRef | Google Scholar
12. Kurano Y, Nakamura M, Ichiba M, Matsuda M, Mizuno E, Kato M, et al. La deficiencia de coreína conduce a una regulación ascendente de la gefirina y el receptor GABA(A). Biochem Biophys Res Commun. (2006) 351:438–42. doi: 10.1016 / j. bbrc.2006.10.070
Resumen de PubMed / Texto Completo de CrossRef / Google Scholar
13. DJ Ehrlich, Walker RH. Neuroimagen funcional y corea: una revisión sistemática. J Clin Mov Disord. (2017) 4:8. doi: 10.1186 / s40734-017-0056-0
Resumen de PubMed / Texto Completo de CrossRef | Google Scholar
14. Connolly BS, Hazrati L-N, Lang AE. Hallazgos neuropatológicos en corea-acanthocytosis: nuevos conocimientos sobre los mecanismos subyacentes de la enfermedad de Parkinson y convulsiones. Acta Neuropatol. (2014) 127:613–5. doi: 10.1007 / s00401-013-1241-3
Resumen de PubMed / Texto Completo de CrossRef | Google Scholar
15. Sorrentino G, De Renzo A, Miniello S, Nori O, Bonavita V. Aparición tardía de acantocitos durante el curso de la corea-acantocitosis. J Neurol Sci. (1999) 163:175–8.
Resumen de PubMed | Google Scholar
16. Bayreuther C, Borg M, Ferrero-Vacher C, Chaussenot A, Lebrun C. Coréo-acantocitosa sin acantocitos. Revue Neurolog. (2010) 166:100–3. doi: 10.1016 / j. neurol.2009.03.005
Texto completo de CrossRef | Google Scholar
17. Lesage S, Drouet V, Majounie E, Deramecourt V, Jacoupy M, Nicolas A, et al. La pérdida de la función VPS13C en el parkinsonismo autosómico recesivo causa disfunción mitocondrial y aumenta la mitofagia dependiente de PINK1 / Parkin. Soy J Hum Genet. (2016) 98:500–13. doi: 10.1016 / j. ajhg.2016.01.014
Resumen de PubMed / Texto Completo de CrossRef / Google Scholar
18. Gauthier J, Meijer IA, Lessel D, Mencacci NE, Krainc D, Hempel M, et al. Las mutaciones recesivas en > VPS13D causan trastornos del movimiento de inicio en la infancia. Ann Neurol. (2018) 83:I6. doi: 10.1002 / ana.25204
Resumen de PubMed | Texto Completo de CrossRef / Google Scholar
19. Kurano Y, Nakamura M, Ichiba M, Matsuda M, Mizuno E, Kato M, et al. Distribución y localización in vivo de la coreína. Biochem Biophys Res Commun. (2007) 353:431–5. doi: 10.1016 / j. bbrc.2006.12.059
Resumen de PubMed | Texto Completo de CrossRef / Google Scholar
20. Lang F, Pelzl L, Schöls L, Hermann A, Föller M, Schäffer TE, et al. Neuronas, eritrocitos y más allá-las diversas funciones de la coreína. Neurosignals (2017) 25: 117-26. doi: 10.1159/000485457
Resumen de PubMed / Texto Completo de CrossRef | Google Scholar
21. Benninger F, Afawi Z, Korczyn AD, Oliver KL, Pendziwiat M, Nakamura M, et al. Convulsiones como síntoma presente y prominente en corea-acantocitosis con mutación del gen c. 2343del VPS13A. Epilepsia (2016) 57: 549-56. doi: 10.1111 / epi.13318
PubMed Abstract | CrossRef Full Text | Google Scholar