Frontiers in Pediatrics
- Introducción
- Síndrome de QT largo
- Las mutaciones del SQTL 1
- SQTL2
- LQTS3
- LQTS4
- SQT5
- LQTS6
- LQTS7
- SQT8
- LQTS9 y LQTS10
- LQTS11
- LQTS12
- LQTS13
- SQTL adquirido
- Las Características electrocardiográficas de las Tres Formas Comunes de Síndrome de QT Largo
- Genotipo-Fenotipo
- El manejo clínico del SQTL
- SQTL: Perspectiva saudita
- Declaración de Conflicto de Intereses
- Agradecimientos
Introducción
Las arritmias cardíacas familiares o hereditarias comprenden porcentajes significativos de arritmias y también causales de muerte cardíaca súbita (MSC) (1, 2). Durante las últimas dos décadas, los científicos y los médicos han realizado un enorme esfuerzo para desentrañar los intrincados y complejos mecanismos de las arritmias familiares congénitas (1-8). Para entender el mecanismo de la arritmogénesis, necesitamos conocer los fundamentos de la estructura celular cardíaca y sus propiedades electrofisiológicas. Los miocitos cardíacos son las principales células que funcionan en el corazón y están ampliamente acoplados para que los impulsos se propaguen rápida y uniformemente. Los cardiomiocitos están separados unos de otros por un límite especializado llamado disco intercalado; las proteínas de unión de separación, los desmosomas cardíacos y los canales iónicos se encuentran en el disco intercalado (9). Las uniones Gap consisten en conexiones compactas que permiten el intercambio intercelular de pequeñas moléculas y también permiten el flujo de las corrientes excitatorias de una célula a su célula vecina. Los desmosomas, junto con las uniones adherentes, son responsables de las uniones mecánicas de los cardiomiocitos individuales. Todos estos componentes en los discos intercalados se segregan secuencialmente y cada componente ejerce su función única; la interrupción de un componente afecta la función de otros componentes, lo que predispone al corazón a desarrollar arritmias (9-11). Los canales iónicos cardíacos son complejos de proteínas formadoras de poros que proporcionan un movimiento interno y externo intrincadamente coordinado y controlado por voltaje de las corrientes iónicas a través de las membranas celulares, esencial para la generación y propagación del ritmo cardíaco. El síndrome de QT largo( SQT), el síndrome de QT corto (SQT), el síndrome del seno enfermo (SSS), el defecto de conducción cardíaca (CCD), el SBR, la taquicardia ventricular polimórfica catecolaminérgica (TVPC), el síndrome de repolarización temprana (ERS) y la Fibrilación Auricular familiar (FA) son las canalopatías cardíacas conocidas actualmente, que podrían ocurrir debido a un defecto único o múltiple en los genes vinculados a la generación y propagación del ritmo cardíaco.
En esta revisión, describiremos exclusivamente las arritmias ligadas al SQTL, su fisiopatología y el manejo clínico disponible en la actualidad. Hemos sido pioneros en dilucidar la patología genética en arritmias cardíacas familiares en Arabia Saudí (1, 12-14), todavía estamos realizando investigaciones cardiogenéticas en Arabia Saudí, al final de esta revisión, discutiremos el conocimiento disponible actualmente sobre los hallazgos genéticos y clínicos obtenidos de varias familias saudíes con antecedentes de síncope y MSC. También añadiremos los datos publicados recientemente sobre SQTL de un equipo del Hospital Especializado y Centro de Investigación Rey Faisal, Riad (15).
Síndrome de QT largo
El SQTL congénito es un trastorno hereditario definido por la prolongación del intervalo QT en el electrocardiograma (ECG). Los pacientes con todas las formas de SQTL están predispuestos a la taquiarritmia ventricular, torsades de pointes (TdP) que conduce a síncope recurrente o MSC. En muchos casos, el síncope o la muerte súbita podrían ser la primera y la única manifestación. Se estima que el SQTL afecta a 1 de cada 2.000 personas en todo el mundo (16). El sello distintivo del SQTL es la prolongación del intervalo QT en el ECG (corregido para la frecuencia cardíaca, es decir, el QTc). Los valores normales de QTc son de 440 ms en hombres y 450 ms en mujeres. En los niños, los valores dependientes de la edad y el género son relevantes. Las recomendaciones de consenso recientes para el diagnóstico de SQTL son las siguientes (17, 18):
1. El SQTL se diagnostica:
a. En presencia de una puntuación de riesgo del SQTL ≥3,5 en ausencia de una causa secundaria de prolongación del intervalo QT y/o
b. En presencia de una mutación patógena inequívoca en uno de los genes del SQTL o
c. En presencia de un intervalo QT corregido para la frecuencia cardíaca utilizando la fórmula de Bazett (QTc) ≥500 ms en electrocardiograma repetido de 12 derivaciones y en ausencia de una causa secundaria para la prolongación del QT.
2. El SQTL puede diagnosticarse en presencia de un QTc entre 480 y 499 ms en ECG repetidos de 12 derivaciones en un paciente con síncope inexplicable en ausencia de una causa secundaria de prolongación del QT y en ausencia de una mutación patógena.
Los pacientes con una duración del intervalo QTc ≥500 ms tuvieron una probabilidad acumulada significativamente mayor de experimentar su primer síncope en comparación con los pacientes con una duración del intervalo QTc <500 ms desde el nacimiento hasta los 20 años de edad (19). Sin embargo, en los pacientes que han experimentado 2, 3 y 4 episodios de síncope, el riesgo de un episodio de síncope posterior fue prácticamente idéntico entre los pacientes que tuvieron una duración de QTc estrecha o prolongada (19). La base molecular del SQTL es heterogénea y, hasta la fecha, se han descrito mutaciones en 13 genes diferentes causales del SQTL (1-8, 19, 20). Por lo general, se encuentra un defecto genético en el 70% de los pacientes con SQTL en uno de estos 13 genes (1-8, 19, 20). Entre los 13 tipos diferentes de SQTL conocidos actualmente, los más comunes son SQTL1, SQTL2 y SQTL3, debido a defectos en los genes del canal iónico cardíaco, KCNQ1, KCNH2 y SCN5A, respectivamente. El noventa por ciento de todas las mutaciones causales del SQTL se encuentran en estos tres genes (1-8, 19, 20). Las mutaciones en los 10 genes restantes son poco frecuentes y comprenden solo 1 10% de todas las mutaciones del SQTL conocidas en la actualidad.
El síndrome de QT largo suele ser una enfermedad autosómica dominante, pero, en ocasiones, se pueden encontrar mutaciones múltiples en un solo gen o en genes diferentes en el 5-10% de los pacientes con SQTL (21, 22). Los pacientes con mutaciones múltiples podrían exhibir un QTc más largo en comparación con aquellos con una sola mutación y dichos pacientes también tienen un riesgo de events 3,5 veces mayor de eventos cardíacos potencialmente mortales (21, 22)
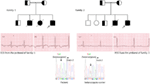
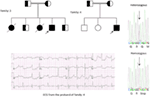
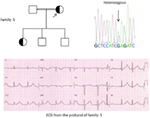
Las mutaciones del SQTL 1
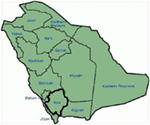
en el gen KCNQ1 son la forma más común de todos los SQTL y se denominan SQTL tipo 1 (SQTL 1). El cincuenta por ciento de todas las mutaciones causales del SQTL se encuentran en el gen KCNQ1 (20). La KvLQT1 (también llamada Kv7. 1) es una proteína producida por el gen KCNQ1 y la proteína forma tetrámeros en el retículo endoplásmico dentro de las células, que supuestamente se ensambla con la proteína Visón (codificada por KCNE1) y luego se transporta a la membrana plasmática de los miocitos cardíacos, donde median una corriente de activación lenta que acelera la repolarización del potencial de acción en los tejidos cardíacos, esta corriente se conoce como IKs (Figura 1). Las mutaciones causales de KCNQ1 del SQT1 son en su mayoría mutaciones sin sentido y, en raras ocasiones, podrían ser mutaciones de desplazamiento de marco en la región C-terminal (23). La arritmia cardíaca en los portadores de la mutación KCNQ1 se desencadena por impulsos adrenérgicos, por ejemplo, estrés emocional, esfuerzo físico, buceo, natación (24-26). Los pacientes con mutaciones en los dominios transmembranarios de KvLQT1 tienen un mayor riesgo de eventos cardíacos relacionados con el SQTL y tienen una mayor sensibilidad a la estimulación simpática (26, 27). Los pacientes con mutaciones sin sentido tienen un mayor riesgo en comparación con los pacientes con mutaciones sin sentido o truncantes (27, 28). La variación en el 3′-UTR en el gen KCNQ1 también afecta significativamente la susceptibilidad a las arritmias, presumiblemente al impactar en la expresión del gen (29). Los pacientes con arritmias debidas a mutaciones en KCNQ1 responden bastante bien a los bloqueadores β, pero algunos pacientes aún podrían ser menos sensibles o incluso resistentes a este medicamento. En un artículo reciente, Barsheshet et al. (30) afirmaron que los pacientes con mutaciones fuera de la región del asa citoplasmática (asa c) en el KvLQT1 son menos sensibles a los bloqueadores β. Figura 1. Corrientes iónicas que contribuyen al potencial de acción ventricular (A) y representación esquemática de un cardiomiocito que muestra (solo) las proteínas involucradas en la patogénesis de los síndromes de arritmias hereditarias (B). En (A), el potencial de acción está alineado con su tiempo aproximado de acción durante el ECG. En (B), no se muestra la anquirina-B, una proteína adaptadora involucrada en el síndrome de QT largo tipo 4. Las mutaciones homocigotas o heterocigotas compuestas en el gen KCNQ1, que podrían causar la forma recesiva de la enfermedad, el síndrome de Jervell y Lange-Nielsen (JLNS), tipo 1 (31), son bastante raras. Los pacientes con SNJ también sufren arritmias cardíacas graves y sordera (31, 32). Los pacientes con mutaciones en KCNQ1 causales de las NJL, por lo general, no tienen ninguna IK funcional (28, 31-33). Es posible que algunos pacientes no tengan sordera a pesar de tener mutaciones homocigotas o heterocigotas compuestas en KCNQ1, estos casos se denominan SQTL autosómicos recesivos 1 (13, 32). En estos pacientes, todavía podría estar presente una pequeña cantidad de corriente IKs funcional (<10% de la IKs total), lo que mantiene la función auditiva, pero sus defectos del ritmo cardíaco son igualmente graves como en los pacientes con NJL (13, 32). Este tipo de SQTL es igualmente prevalente que el SQTL1, representando el 35-40% de los pacientes con SQTL con una mutación detectable (1, 20, 24, 25). KCNH2 codifica para la proteína HERG (Kv11.1), que es la subunidad α del rectificador retardado de rápida activación K+ actual (IKr). Las mutaciones patógenas en este gen que reducen la función del canal Kv11.1 prolongan la duración del intervalo QT (Figura 1) y son causales del SQTL 2. El veintinueve por ciento de los ataques sincopales en el SQTL 2 ocurren durante el descanso/sueño y solo el 13% de los ataques sincopales se reportaron durante el ejercicio (25, 34). Ruidos repentinos y sorprendentes, por ejemplo, timbres de despertador, timbres de puerta, timbres de teléfono, suelen desencadenar ataques sincopales en estos pacientes (24, 34). Los pacientes con mutaciones en la región formadora de poros del Kv11.1 (codificada por el gen KCNH2) son susceptibles a un riesgo alto de eventos cardíacos relacionados con arritmias en comparación con los pacientes con mutaciones en la región no porosa (35). En neonatos, el bloqueo auriculoventricular (AV) 2:1 se asocia preferentemente con mutaciones en KCNH2 (36). El bloqueo AV completo complicado por SQTL también se encontró en el 17% de los pacientes adultos con una mutación en el gen KCNH2 (37). Las mutaciones homocigotas en KCNH2 son raras y, cuando están presentes, los pacientes sufren una forma grave de SQTL, con 2:1 Bloqueo AV y arritmias ventriculares graves, tanto durante las etapas intrauterinas como después del nacimiento (11, 38-40). Además de las numerosas mutaciones descritas en la patología del SQTL 2, las variantes polimórficas comunes en el gen KCNH2 también podrían modular la gravedad de la enfermedad. Un ejemplo interesante es el polimorfismo K897T (SNP) en KCNH2, que está presente en el 33% de la población general (41). Se notificó que K897T exacerbaba la patogenicidad de las mutaciones de KCNH2 (42, 43). Nav1.5 es la subunidad α formadora de poros del canal Na+ cardíaco dependiente de voltaje, es una proteína de membrana integral codificada por el gen SCN5A y está involucrada en la iniciación y conducción de potenciales de acción cardíacos. Los canales cardiacos Na+ están compuestos de una subunidad α formadora de poros (codificada por SCN5A) y una o más subunidades β auxiliares. El SQTL 3 es causado por mutaciones de ganancia de función que interrumpen la inactivación rápida de la subunidad α (Figura 1) y se ha descrito una mutación en el gen SCN5A en <10% de todos los pacientes con SQTL con una mutación (1, 20, 24). Los pacientes con SQTL 3 experimentan la mayoría (39%) de sus eventos cardíacos durante el sueño/descanso (25), y alrededor de ∼13% de los eventos ocurrieron durante el ejercicio (25). En varios casos, una sola mutación de SCN5A demostró ejercer dos o incluso tres fenotipos distintos de arritmias en la misma familia, por ejemplo, SQTL, SBR o CCD (44-47). Los pacientes varones con una mutación en el SQTL 3 podrían presentar síntomas mucho antes que las mujeres (48). Se han descrito mutaciones en SCN5A heterocigotas y homocigotas en el SQTL3 con bloqueo AV funcional 2:1 (49). Recientemente hemos encontrado una familia iraní con la mutación 1507_1509delQKP en varios miembros de la familia, donde los pacientes tenían SQTL combinado y CCD (datos no mostrados). Esta mutación también se ha notificado en pacientes de otros países, lo que sugiere que se trata de una mutación recurrente y de punto caliente . También se notificaron mutaciones con tales características de pérdida y ganancia de función durante diferentes fases del potencial de acción en las mutaciones de 1493delK y 1795insD (44, 51). Ocasionalmente, un SNP también podría ejercer un efecto patógeno en sus portadores. S1103Y es una variante común del gen SCN5A, presente en el 13% de los afroamericanos (52). Los portadores con esta variante tienen un mayor riesgo de arritmias y síndrome de muerte súbita del lactante (SMSL) (52). LQTS4 representa la primera forma de LQTS sin canal. Una mutación en el ANK2, una proteína adaptadora, conduce a una sobrecarga de calcio intracelular que contribuye al SQTL 4 (53, 54). Además de la prolongación del intervalo QT, los pacientes con este síndrome podrían tener bradicardia sinusal, FA paroxística y TVPC (54). El efecto patógeno de las mutaciones en ANK2 puede ser de moderado a grave y las expresiones clínicas dependen de la gravedad de la mutación. Las mutaciones en KCNE1 se asocian con el SQT5 (Figura 1) (55, 56). Las mutaciones heterocigotas de KCNE1 reducen la IKs al ejercer un efecto negativo dominante sobre el alelo normal que lo acompaña, y provocan un retraso en la repolarización cardíaca (Figura 1), responsable del aumento del riesgo de arritmias (6). Los pacientes con mutaciones homocigotas en KCNE1 sufren de NJL (tipo 2) (57, 58). D85N es un polimorfismo en el gen KCNE1, presente en 0.7-1% de la población general (41). En un estudio de Nishio et al. (59), el polimorfismo D85N se encontró con mayor frecuencia en los pacientes con SQTL, lo que lo convierte en un genotipo de riesgo en la patología del SQTL (potencialmente solo en la población asiática). En Europa, se notificó D85N en el 5% de los pacientes con SQTL adquirido (SQTL) (en una cohorte de 32 pacientes) que habían experimentado TdP (60). El gen KCNE2 codifica para el péptido relacionado con el visón 1 (MiRP1), una supuesta subunidad β del canal de potasio cardíaco IKr (Figura 1). Las mutaciones en el gen KCNE2 también podrían conducir a defectos en el componente de activación rápida de la corriente de potasio rectificadora retardada (IKr), base patológica del SQTL 6 (61). Estímulo auditivo / acústico como ruido de despertador, timbre, etc. podría provocar ataques sincopales en portadores de la mutación KCNE2, similar a la mutación KCNH2 (62). Este síndrome también se conoce como síndrome de Andersen-Tawil (ATS). El STA es un trastorno poco frecuente, que se manifiesta por síncope ocasional y paro cardíaco. Las características del ECG incluyen prolongación leve del intervalo QT, ondas U anormales, ectopía ventricular frecuente, taquicardia ventricular bidireccional (TV) y TV polimórfica. Este síndrome también presenta características extracardíacas, por ejemplo, parálisis periódica del músculo esquelético y problemas de desarrollo, como paladar hendido, orejas bajas, estatura baja y defectos de desarrollo en las extremidades (63). Se informó que la mayoría de los pacientes con STA diagnosticados clínicamente tenían una mutación en KCNJ2 (63). KCNJ2 codifica una subunidad formadora de poros de canales de potasio rectificadores internos (IK1) (Figura 1) (64, 65). También conocido como síndrome de Timoteo (ST), los pacientes muestran una prolongación severa del intervalo QT en sus ECG, que se combina con sindactilia, calvicie al nacer y dientes pequeños en el 100% de los casos y malformaciones estructurales cardíacas menos penetrantes, retraso mental, autismo y características dismórficas faciales (66). Hay dos subtipos: TS1 (clásico) y TS2 (forma rara). La TS2 es cardiológicamente más severa que la TS1 (66, 67). Los pacientes con TS2 también carecen de sindactilia (67). Las mutaciones en la subunidad α-1 de la corriente de calcio de tipo L (CAC-L) que codifica el gen CACNA1C conducen a ambas formas de TS (LQTS8). En el corazón y el cerebro, donde el CACNA1C se expresa predominantemente, el exón-8 se encuentra en 8 el 80% de las transcripciones del ARNm y el exón-8A está presente transc el 20% de las transcripciones (66). La mutación G406R en el exón-8A es causal a la forma clásica de TS (TS1) y se notificó G402S en el exón-8 en el más severo en TS2. Una nueva adición a esta lista es la mutación Ala1473Gly, que se ha descrito en un bebé con síndrome de ST con fenotipo expandido adicional (68). Todas las mutaciones fueron de novo o mosaico en el progenitor, y todas las ganancias resultantes de la función del canala-L (66-69). Las mutaciones en CAV3 o SCN4B producen ganancia de función en el INa tardío, causando un fenotipo similar al LQTS3 (70-74). Se conocen como LQTS9 (asociado con la mutación CAV3) y LQTS10 (asociado con la mutación SCN4B). Las caveolas están bien descritas por Engelman et al. (72) como “pequeñas cuevas” en la membrana plasmática. Son pequeñas fosas sin recubrimiento y se consideran el lugar de importantes eventos dinámicos y reguladores en la membrana plasmática (72, 73). Las caveolinas son las principales proteínas en las caveolas, y la caveolina-3 (codificada por el gen CAV3) se encuentra específicamente en los cardiomiocitos y las células musculares esqueléticas. Se ha informado específicamente que varios canales iónicos cardíacos están localizados en las caveolas extraídas de miocitos cardíacos enriquecidos en caveolina-3 (72, 73). Además, los componentes de la cascada de señalización del receptor β-adrenérgico también están presentes en membranas enriquecidas con caveolas (72, 73). SCN4B codifica para NaVß4, que es una subunidad β auxiliar del canal de sodio cardíaco. Hasta el momento, solo se ha reportado una mutación (L179F) en este gen en una familia mexicana con múltiples miembros afectados (74). La mutación en este gen resultó en una ganancia de función de la corriente Nav1.5 (74). En el corazón, la regulación simpática de la duración del potencial de acción cardíaco (DPA) está mediada por la activación del receptor β-adrenérgico (β-AR), que requiere el ensamblaje de AKAP9 (Yotiao) con la subunidad α (KvLQT1) del canal IKs. La mutación en AKAP9 causa SQTL 11 (75). Hasta la fecha, solo se ha notificado una mutación, S1570L, en AKAP9 (75). La mutación en el gen α-1-Sintrofina (SNTN1) es causal de LQTS12. La mutación en este gen conduce a la ganancia de función del canal de sodio cardíaco (Nav1.5), que es la base patológica del SQTL12 (76). La subunidad del canal de potasio rectificadora interna acoplada a proteínas G (Kir3.4) está codificada por el gen KCNJ5. Una mutación de pérdida de función en este gen podría causar SQTL 13 (77). Hasta ahora, solo se ha descrito una mutación, G387R, en una familia china con nueve pacientes con esta mutación. Se sugirió una expresión reducida de Kir3.4 en la membrana plasmática como patología del SQTL en los pacientes. Además del SQTL congénito, también existe otra variante de SQTL conocida como SQTL, que es causada por factores y sustancias que disminuyen el flujo de potasio y perjudican la capacidad del miocardio para repolarizarse. Las afecciones bien reconocidas son el género femenino, la hipopotasemia y los medicamentos que inhiben los canales cardíacos de potasio (78-80). Una serie de medicamentos recetados comúnmente también podrían unirse y bloquear preferentemente el canal HERG (Kv11.1, una proteína codificada por el gen KCNH2) y predisponer al aLQTS (79, 80). Recientemente, se demostró que el bloqueo de los SQi también podría contribuir a los ALQT inducidos por fármacos, especialmente cuando la reserva de repolarización está comprometida (81). Se encontró que la fluoxetina y la norfluoxetina suprimían las propiedades de la IKs, tanto in vivo como in vitro, y dieron lugar a SQTL marcados (81). Polimorfismos, D85N en visón (gen KCNE1), T8A, Q9E en MiRP1 (gen KCNE2), que son supuestas subunidades β de los canales IKs e IKr, fueron reportados para causar ALQT (82, 83). También se ha notificado SQTL autoinmune en un paciente con anticuerpos anti-HERG que contienen IgG (84). Los patrones típicos de onda T-St están presentes en la mayoría de los pacientes con SQTL genotipado y se pueden utilizar para identificar los genotipos SQT1, SQT2 y posiblemente SQT3 (85, 86). La forma del SQTL 1 del SQTL se asocia a una onda T amplia sin acortar el intervalo QT en el ejercicio (Figura 2A). El SQTL 2 se asocia con ondas T de baja amplitud, a menudo bífidas (Figura 2B). El LQTS3 está asociado con un segmento largo iso-eléctrico y una onda T alta de base estrecha (Figura 2C). La dependencia de pausa del inicio de TdP en el SQTL congénito es específica para el genotipo, siendo predominante en el SQTL2 pero casi ausente en el SQTL1 (87). Figura 2. Registros electrocardiográficos de pacientes con SQTL 1, SQTL 2 y SQTL 3. A) ECG de 12 derivaciones de un varón de 18 años con una mutación KCNQ1. El intervalo QT se prolonga (QTc = ±500 ms). El segmento ST tiene una base amplia y una amplitud relativamente grande. El intervalo de conducción es normal (calibración estándar). B) ECG de 12 derivaciones de una niña de 14 años con una mutación en KCNH2. El intervalo QT se prolonga (QTc ± 520 ms). El segmento ST tiene muescas en la derivación V3 y tiene una amplitud relativamente baja en las derivaciones de las extremidades. El intervalo de conducción es normal (calibración estándar). C) ECG de 12 derivaciones de un niño de 12 años con una mutación en el SCN5A. El intervalo QT se prolonga (QTc ± 600 ms). El segmento ST tiene un segmento largo (casi) iso-eléctrico con una onda T grande, afilada y estrecha. El intervalo de conducción es normal (calibración estándar). Aunque los patrones pueden sugerir un genotipo específico del SQTL, se han encontrado con frecuencia excepciones. El síndrome de QT largo es una enfermedad autosómica dominante. El análisis de genotipos y fenotipos entre los portadores de mutaciones heterocigotas se ha llevado a cabo ampliamente en los pacientes con SQTL 1, SQTL 2 y SQTL 3. Durante la infancia, el riesgo de eventos cardíacos es significativamente mayor en los varones con SQTL 1 que en las mujeres con SQTL 1, mientras que no se observaron diferencias significativas relacionadas con el género en el riesgo de eventos cardíacos entre los pacientes con SQTL 2 y SQTL 3 (88-91). Durante la edad adulta (también después de los 40 años), las mujeres con SQTL 1 y SQTL 2 podrían tener un riesgo significativamente mayor de eventos cardíacos que los hombres respectivos (88-91). En general, la letalidad de los eventos cardíacos parece ser predominante en los pacientes con SQTL 3 que en los pacientes con SQTL 1 y SQTL 2 (91). Las mujeres con SQTL tienen un riesgo reducido de eventos cardíacos durante el embarazo, pero el riesgo aumenta bastante durante el período posparto de 9 meses, especialmente en las mujeres con mutación en el gen KCNH2 (92). La muerte súbita cardíaca en niños también podría ser causada por mutaciones en los genes del canal iónico cardíaco (93-96). Aproximadamente el 28% de los niños con una MSC inexplicable (entre 1 y 18 años, edad media: 12,3 ± 3,8 años) eran portadores de mutaciones en los genes causales del SQTL (97). En el SMSL, las mutaciones en el SCN5A parecían predominantes (98, 99), pero también se encontraron mutaciones en KCNQ1, KCNH2, KCNE2 y CAV3, SCN4B y SCN3B (100, 101). También se notificaron muertes fetales intrauterinas debido a defectos en los genes del canal iónico cardíaco (12, 102). El cese de todos los fármacos que se sabe que prolongan el intervalo QT y también la corrección de los desequilibrios electrolíticos y/o las condiciones metabólicas precipitantes deben ser el enfoque principal al tratar a los pacientes con SQTL (adquiridos). Los síntomas en el SQTL a menudo están mediados por vía adrenérgica, por lo que generalmente se recomienda restringir la participación de los pacientes en actividades deportivas (24, 25). El pilar de la terapia clínica para el SQTL es el bloqueo β. Generalmente se usan preparaciones de acción prolongada de propranolol, nadolol y metoprolol, y su eficacia en el bloqueo β se evalúa mediante el embotamiento de la frecuencia cardíaca de ejercicio (por ejemplo, mediante >20%) (24, 25). Entre todos los bloqueadores β, el propranolol y el nadolol se consideran superiores al metoprolol en pacientes sintomáticos (103). Además, el bloqueo β también se puede utilizar como tratamiento profiláctico en portadores de mutaciones silenciosas para reducir la MSC (24). En un estudio de Barsheshet et al. (30), los pacientes con mutaciones sin sentido del asa c en el gen KCNQ1 mostraron un alto riesgo de eventos cardíacos potencialmente mortales y obtuvieron beneficios significativos del tratamiento con bloqueadores β. Como las mujeres con SQTL 2 tienen un mayor riesgo durante el período posparto de 9 meses, se deben recetar bloqueadores β para reducir cualquier evento cardíaco durante este período de alto riesgo (92). Se puede considerar un desfibrilador automático implantable (DAI) para pacientes con síncope recurrente a pesar de la terapia con bloqueadores β o en pacientes con alto riesgo de paro cardíaco (p. ej., SQTL sintomático 2 y SQTL 3 con prolongación documentada del intervalo QTc). La denervación simpática cardíaca izquierda (DSCC) se recomienda para pacientes con SQTL de alto riesgo en los que un DAI está contraindicado o rechazado y los bloqueadores β no son eficaces, no se toleran, no se aceptan o están contraindicados (18, 104). Los pacientes con NJL suelen tener un QTc > 500 ms y también tienen un alto riesgo, los bloqueadores β no tienen eficacia suficiente en aquellos pacientes en los que se recomendó un tratamiento temprano con DAI (18, 31). El primer informe sobre el SQTL de Arabia Saudita se publicó en 1993 en el Hospital de las Fuerzas Armadas de Riad (105). Cuatro bebés y niños pequeños de entre 6 y 48 meses con antecedentes de convulsiones recurrentes, de una sola familia, fueron diagnosticados como SQTL (105). Los antecedentes familiares revelaron que otros dos miembros de la familia extendida tuvieron episodios similares de pérdida repentina del conocimiento y tres miembros de la familia murieron repentinamente (105). En todos los casos, el diagnóstico inicial fue epilepsia (105). Varios años después, se notificaron dos casos esporádicos con variantes comparativamente más severas de SQTL neonatal combinadas con bloqueo AV 2:1 (106, 107). Todos estos informes clínicos publicados carecían de hallazgos genéticos que pudieran explicar la fisiopatología del SQTL en ellos (105-107). Por primera vez, hemos reportado defectos genéticos como base patológica del SQTL en pacientes similares de Arabia Saudita. Hemos investigado a seis familias saudíes con antecedentes de síncope y muertes repentinas e inexplicables de feto, neonatos y niños (12-14). El SQTL autosómico recesivo 1 se diagnosticó en niños de dos familias (Figura 3). El SQTL autosómico recesivo 2 se diagnosticó en dos familias (Figura 4). En una familia, una paciente de sexo femenino fue diagnosticada con SQTL autosómico dominante 2 (Figura 5), la paciente tuvo ataques sincopales durante el período de recuperación posparto en el hospital, lo cual es muy común en mujeres portadoras de la mutación KCNH2. En todos nuestros pacientes, la identificación de mutaciones patógenas en los genes causales del canal iónico cardíaco del SQTL llevó a un diagnóstico clínico confirmado, que se diagnosticaron erróneamente como convulsiones epilépticas antes de ser remitidos a nosotros (13, 14) Recientemente, Shinwari et al. (15) del King Faisal Specialist Hospital and Research Centre informó de una mutación causal de KCNQ1 en el LQTS1, H258P, en una familia numerosa con 12 individuos afectados. Solo dos portadores eran sintomáticos, su QTc > 500 ms, y los bloqueadores β suprimieron los síntomas clínicos en un paciente y el segundo paciente sintomático requirió un DAI. Gráfico 3 Dibujo de pedigrí de la familia-1 y 2 con SQTL autosómico recesivo 1, los probandos se muestran con una flecha. Los ECG de los probandos en dos familias se muestran en el medio, con un QTc de 557 y 529 ms, respectivamente. Se encontró mutación intrónica, c. 387 -5 T > A en el gen KCNQ1 en los pacientes (mostrado en la parte inferior). La mutación se mostró con una flecha. Los círculos y cuadrados sin relleno no son portadores de la mutación. Los individuos afectados se muestran en círculos llenos (mujeres) y cuadrados (hombres). Los cuadrados y círculos a medio llenar son individuos con mutación heterocigota. Los individuos fallecidos se indican con barras, los probandos se indican con una flecha y el matrimonio consanguíneo se indica con = El límite Exón − intrón se muestra con una línea de puntos con una flecha que apunta hacia el exón. Figura 4. Dibujo de pedigrí de la familia-3 y 4 con SQTL autosómico recesivo 2. El ECG del probanda de la familia-4 se muestra debajo del dibujo pedigrí, que muestra taquicardia sinusal, bloqueo AV casi completo, ritmo de escape amplio y complejo con intervalos QTc muy largos (QT >600 ms). Se encontró mutación, c. 3208 C > T (p. Q1070X) en el gen KCNH2 en los pacientes (se muestra a la derecha), marcada con una flecha. Figura 5. Arriba a la izquierda: pedigrí de la familia-5. El individuo afectado se muestra con un círculo lleno (hembra) El Proband se indica con una flecha. ECG de 12 derivaciones del probanda (I:2). El ECG muestra una onda T de gran amplitud, de base amplia y pico alto, el intervalo QTc es de 580 ms. La detección del gen KCNH2 muestra la sustitución del nucleótido “G” por un “A” (c. 2362G > A, marcado con una flecha), lo que conduce a la sustitución de aminoácidos, p. E788K. Los hallazgos genéticos y clínicos de nuestro estudio (12-14) de Arabia Saudita son bastante intrigantes por varias razones: (1) En total, hemos investigado seis familias, entre ellas cuatro eran homocigotas/heterocigotas compuestas para las mutaciones y las mutaciones se originaron de una fuente ancestral; (2) Todas las mutaciones en SQTL fueron nuevas, reportadas solo en estas familias árabes; (3) Debido a la homocigosidad o heterocigosidad compuesta para las mutaciones, los fenotipos clínicos también fueron severos en nuestras familias estudiadas (12-14). Sugerimos que las observaciones genéticas y fenotípicas se derivaban de la tasa extremadamente alta de matrimonios consanguíneos en Arabia Saudita (108, 109). Nuestro estudio proporcionó la primera evidencia científica sobre el papel de la consanguinidad en el ejercicio de un papel fundamental en las arritmias cardíacas inexplicables y las SCD en niños y adolescentes en Arabia Saudí (12-14). También hemos demostrado que la mutación KCNQ1 c. 387 -5 T > A (NM_000218) (Figura 3) se había propagado en la provincia de Assir de Arabia Saudita a partir de un ancestro común durante varias generaciones debido a la alta incidencia de matrimonios consanguíneos . Hasta el momento, esta es la mutación causal del SQT1 más común en la población de Arabia Saudita, que también se observó en el Hospital Especializado Rey Faisal y en el Hospital Militar Khamis Mashayt (sin publicar). También se obtuvo un resultado similar para la mutación causal del SQT2, c. 3208 C > T (p.Q1070X) (Figura 4) en el gen KCNH2 (12, 14), que también es una mutación fundadora en Arabia Saudita y segregada durante muchas generaciones en la región de Assir (ver el mapa, Figura 6). Predecimos que existe un número considerable de individuos con las mutaciones ancestrales/fundadoras mencionadas (tanto en KCNQ1 como en KCNH2 y otros genes) en esta región y también en las grandes ciudades como Riad, Jeddah y Dammam debido a la migración urbana. En otras provincias de Arabia Saudita se prevén más mutaciones fundadoras o ancestrales patógenas para el SQTL debido a la alta tasa de matrimonios consanguíneos. Gráfico 6 Mapa de Arabia Saudita (cortesía de Wikipedia). La región de Assir está marcada con una línea gruesa, donde hemos encontrado las mutaciones fundadoras en los genes KCNQ1 y KCNH2, descritas en la familia 1-4. Como el SQTL es una enfermedad autosómica dominante, esperábamos observar pacientes portadores de mutaciones predominantemente heterocigotas. Pero en nuestro estudio, identificamos principalmente pacientes con mutaciones recesivas (12-14). Claramente, estos pacientes se convierten en primeros sintomáticos y, fueron remitidos predominantemente al Centro Cardíaco Príncipe Sultán, poniéndolos bajo nuestra atención. Sin embargo, los hallazgos de nuestras investigaciones implican que el SQTL recesivo podría tener fenotipos clínicos mortales en niños, y no son infrecuentes en Arabia Saudí (12-14). Como los portadores de mutaciones heterocigotas también son susceptibles de desarrollar arritmias y sus complicaciones, se debe tomar una iniciativa concertada para reunir a los médicos generales, cardiólogos y genetistas clínicos locales en una plataforma común para identificar a los individuos en riesgo. Además, por el momento, tampoco tenemos información genética para otros trastornos de arritmias familiares, por ejemplo, TVPC, SQT, BrS, AF, etc. Los centros cardiogenéticos especializados deben tomar la iniciativa de buscar defectos genéticos, mutaciones y realizar estudios genotipo-fenotipo en todas las formas de arritmias hereditarias. También debe tenerse en cuenta que no todas las arritmias genéticas tendrían antecedentes familiares, ya que en muchos casos la mutación causal de la arritmia tiene un origen de novo, es decir, el probanda es el primer paciente de esa familia con la mutación y es la fuente de transmisión de la mutación en generaciones posteriores (110, 111). Debido a la muy alta tasa de matrimonios consanguíneos en Arabia Saudita, esperamos que muchas más mutaciones del fundador ejerzan un papel crucial en las arritmias congénitas en este país. Identificar estas mutaciones fundadoras debería ser nuestra primera y principal tarea, lo que nos facilitaría el desarrollo de un asesoramiento genético prematrimonial y también pre-sintomático efectivo en este país. Las mutaciones en los genes causales del SQTL podrían conferir variabilidad en la penetrancia clínica, en un extremo, algunos portadores podrían presumiblemente estar completamente sanos, pero algunos portadores podrían tener su primera manifestación de la enfermedad como síncope o muerte súbita. La muerte súbita de un niño pequeño o un adulto representa una gran carga psicológica y emocional para la familia, la detección de los portadores es esencial, ya que existe un medicamento simple, por ejemplo, bloqueadores β (también modificación del comportamiento), que podría prevenir de manera muy efectiva a los portadores de las consecuencias fatales de la arritmia y las SCD. Los individuos con mutaciones homocigotas en los genes causales del SQTL podrían tener una morbilidad grave y una tasa de mortalidad extremadamente alta, incluidas las bradiarritmias fetales y, en muchos casos, abortos espontáneos en las madres embarazadas (12-14). No se ha realizado mucha investigación en Arabia Saudita sobre la prevalencia de varios SNP en los genes vinculados a arritmias y también en los genes que los regulan. Splawski et al. (112) describieron una variante común de S1103Y en el gen SCN5A asociada con arritmia en afroamericanos. El alelo variante denominado Y1103 es responsable de acelerar la activación del canal, aumentando así la probabilidad de arritmias cardíacas en personas de ascendencia africana (52, 112). K393N es una variante en el gen KCNQ1 reportada en pacientes con SQTL 1 en EE.UU., pero en la población árabe hemos detectado esta variante en el 2% de los individuos (datos no publicados). Si la variante K393N en el gen KCNQ1 en árabes es comparable a la variante S1103Y (SCN5A) en afroamericanos o la variante D85N (KCNE1) en la población japonesa también podría merecer investigación (59). Los autores declaran que la investigación se realizó en ausencia de relaciones comerciales o financieras que pudieran interpretarse como un conflicto de intereses potencial. Nuestro sincero agradecimiento al Prof. Arthur Wilde, al Prof. Connie Bezzina y al editor de la revista “Heart” por su permiso para usar la Figura 1 en este manuscrito de la Ref. (113). 1. Bhuiyan ZA. Espectro Clínico y Genético de Síndromes de Arritmias Cardíacas Hereditarias. Amsterdam: Amsterdam University (2009). 2. Priori SG, Aliot E, Blømstrom-Lundqvist C, Bossaert L, Breithardt G, Brugada P, et al. Grupo de trabajo sobre muerte súbita cardíaca, sociedad europea de cardiología. Europace (2002) 4: 3-18. doi: 10.1053 / eupc.2001.0214 Texto completo cruzado 3. Wang Q, Shen J, Splawski I, Atkinson D, Li Z, Robinson JL, et al. Mutaciones en SCN5A asociadas con una arritmia cardíaca hereditaria, síndrome de QT largo. Cell (1995) 80: 805-11. doi:10.1016/0092-8674(95)90359-3 Resumen de Pubmed / Texto Completo de Pubmed / Texto Completo de CrossRef 4. Curran ME, Splawski I, Timothy KW, Vincent GM, Green ED, Keating MT. Una base molecular para la arritmia cardíaca: las mutaciones HERG causan el síndrome de QT largo. Cell (1995) 80: 795-803. doi: 10.1016/0092-8674(95)90358-5 Resumen de Pubmed / Texto Completo de Pubmed / Texto Completo de CrossRef 5. Wang Q, Curran ME, Splawski I, Burn TC, Millholland JM, VanRaay TJ, et al. Clonación posicional de un nuevo gen del canal de potasio: las mutaciones en KVLQT1 causan arritmias cardíacas. Nat Genet (1996) 12:17-23. doi: 10.1038 / ng0196-17 Resumen de Pubmed / Texto Completo de Pubmed / Texto Completo de CrossRef 6. Splawski I, Tristani-Firouzi M, Lehmann MH, Sanguinetti MC, Keating MT. Las mutaciones en el gen hminK causan el síndrome de QT largo y suprimen la función IKs. Nat Genet (1997) 17:338-40. doi: 10.1038 / ng1197-338 Resumen de Pubmed / Texto Completo de Pubmed / Texto Completo de CrossRef 7. Sanguinetti MC, Jiang C, Curran ME, Keating MT. Un vínculo mecánico entre una arritmia cardíaca heredada y una adquirida: HERG codifica el canal de potasio IKr. Cell (1995) 81: 299-307. doi:10.1016/0092-8674(95)90340-2 Resumen de Pubmed / Texto Completo de Pubmed / Texto Completo de CrossRef 8. Neyroud N, Tesson F, Denjoy I, Leibovici M, Donger C, Barhanin J, et al. Una nueva mutación en el gen del canal de potasio KVLQT1 causa el síndrome cardioauditorio de Jervell y Lange-Nielsen. Nat Genet (1997) 15:186-9. doi: 10.1038 / ng0297-186 Resumen de Pubmed / Texto Completo de Pubmed / Texto Completo de CrossRef 9. Kucera JP, Rohr S, Rudy Y. Localización de canales de sodio en discos intercalados modula la conducción cardíaca. Circ Res (2002) 91: 1176-82. doi: 10.1161 / 01.RES.0000046237.54156.0 Pubmed Abstract | Pubmed Texto Completo | CrossRef Texto Completo 10. Oxford EM, Musa H, Maass K, Coombs W, Taffet SM, Delmar M. Remodelación de Connexin43 causada por la inhibición de la expresión de plakophilin-2 en las células cardíacas. Circ Res (2007) 101:703-11. doi: 10.1161 / CIRCRESAHA.107.154252 Resumen de Pubmed / Texto Completo de Pubmed / Texto Cruzado Completo 11. Rohr S. Diafonía molecular entre uniones mecánicas y eléctricas en el disco intercalado. Circ Res (2007) 101:637-9. doi: 10.1161 / CIRCRESAHA.107.161901 Texto completo del texto cruzado 12. Bhuiyan TS, Momenah TS, Gong Q, Amin AS, Ghamdi SA, Carvalho JS, et al. Pérdida fetal intrauterina recurrente debido a la casi ausencia de HERG: caracterización clínica y funcional de una mutación homocigótica sin sentido HERG Q1070X. Heart Rhythm (2008) 5: 553-61. doi: 10.1016 / j. hrthm.2008.01.020 Resumen de Pubmed / Texto Completo de Pubmed / Texto Cruzado Completo 13. Bhuiyan TS, Momenah TS, Amin TS, Al-Khadra AS, Alders M, Wilde AAM, et al. Una mutación intrónica que conduce a la omisión incompleta del exón-2 en KCNQ1 rescata la audición en el síndrome de Jervell y Lange-Nielsen. Prog Biophys Mol Biol (2008) 98: 319-27. doi: 10.1016 / j. pbiomolbio.2008.10.004 Resumen de Pubmed / Texto Completo de Pubmed / Texto Cruzado Completo 14. BhuiyanA, Al-Shahrani S, Al-Khadra AS, Al-Ghamdi S, Al-Kalaf K, Mannens MMAM, et al. Análisis clínico y genético del síndrome de QT largo en niños de seis familias en Arabia Saudita: ¿son diferentes? Pediatr Cardiol (2009) 30:490-501. doi: 10.1007 / s00246-008-9377-y Resumen de Pubmed / Texto Completo de Pubmed / Texto Cruzado Completo 15. Shinwari ZM, Al-Hazzani A, Dzimiri N, Tulbah S, Mallawi Y, Al-Fayyadh M, et al. Identification of a novel KCNQ1 mutation in a large Saudi family with long QT syndrome: clinical consequences and preventive implications (en inglés). Clin Genet (2013) 83:370-4. doi: 10.1111 / j. 1399-0004. 2012. 01914.x Resumen de Pubmed / Texto Completo de Pubmed / Texto Cruzado Completo 16. Schwartz PJ, Stramba-Badiale M, Crotti L, Pedrazzini M, Besana A, Bosi G, et al. Prevalencia del síndrome congénito de QT largo. Circulation (2009) 120:1761-7. doi: 10.1161 / CIRCULATIONAHA.109.863209 Resumen de Pubmed / Texto Completo de Pubmed / Texto Cruzado Completo 17. Schwartz PJ, Moss AJ, Vincent GM, Crampton RS. Criterios diagnósticos para el síndrome de QT largo. Una actualización. Circulation (1993) 88: 782-4. doi: 10.1161 / 01.CIR.88.2.782 Texto completo cruzado 18. Priori SG, Wilde AA, Horie M, Cho Y, Behr ER, Berul C, et al. Resumen ejecutivo: Declaración de consenso de HRS/EHRA/APHRS sobre el diagnóstico y el manejo de pacientes con síndromes de arritmias primarias hereditarias. Europace (2013) 15:1389-406. doi: 10.1093/europace / eut272 Texto completo cruzado 19. Liu JF, Jons C, Moss AJ, McNitt S, Peterson DR, Qi M, et al. Registro de Síndrome. Factores de riesgo de síncope recurrente y acontecimientos posteriores mortales o casi mortales en niños y adolescentes con síndrome de QT largo. J Am Coll Cardiol (2011) 57:941-50. doi: 10.1016 / j. jacc.2010.10.025 Resumen de Pubmed / Texto Completo de Pubmed / Texto Cruzado Completo 20. Splawski I, Shen J, Timothy KW, Lehmann MH, Priori S, Robinson JL, et al. Espectro de mutaciones en genes del síndrome de QT largo. KVLQT1, HERG, SCN5A, KCNE1 y KCNE2. Circulation (2000) 102:1178-85. doi: 10.1161 / 01.CIR.102.10.1178 Resumen de Pubmed / Texto Completo de Pubmed / Texto Cruzado Completo 21. Westenskow P, Splawski I, Timothy KW, Keating MT, Sanguinetti MC. Mutaciones compuestas: una causa común del síndrome de QT largo grave. Circulation (2004) 109:1834-41. doi: 10.1161 / 01.CIR.0000125524.34234.13 Resumen de Pubmed / Texto Completo de Pubmed / Texto Cruzado Completo 22. Mullally J, Goldenberg I, Moss AJ, Lopes CM, Ackerman MJ, Zareba W, et al. Riesgo de eventos cardíacos potencialmente mortales en pacientes con síndrome de QT largo y mutaciones múltiples. Heart Rhythm (2013) 10:378-82. doi: 10.1016 / j. hrthm.2012.11.006 Resumen de Pubmed / Texto Completo de Pubmed / Texto Cruzado Completo 23. Napolitano C, Priori SG, Schwartz PJ, Bloise R, Ronchetti E, Nastoli J, et al. Pruebas genéticas en el síndrome de QT largo: desarrollo y validación de un enfoque eficiente del genotipado en la práctica clínica. JAMA (2005) 294:2975-80. doi: 10.1001 / jama.294.23.2975 Resumen de Pubmed / Texto Completo de Pubmed / Texto Cruzado Completo 24. Roden DM. Práctica clínica. Síndrome de QT largo. N Engl J Med (2008) 358:169-76. doi: 10.1056 / NEJMcp0706513 Texto completo cruzado 25. Schwartz PJ, Priori SG, Spazzolini C, Moss AJ, Vincent GM, Napolitano C, et al. Correlación genotipo-fenotipo en el síndrome de QT largo: desencadenantes específicos de genes para arritmias potencialmente mortales. Circulation (2001) 103:89-95. doi: 10.1161 / 01.CIR.103.1.89 Texto completo cruzado 26. Ackerman MJ, Tester DJ, Porter CJ. Natación, un desencadenante arritmogénico específico de un gen para el síndrome hereditario de QT largo. Mayo Clin Proc (1999) 74: 1088-94. doi:10.4065/74.11.1088 Resumen de Pubmed / Texto Completo de Pubmed / Texto Completo de CrossRef 27. Kapa S, Tester DJ, Salisbury BA, Harris-Kerr C, Pungliya MS, Alders M, et al. Pruebas genéticas para el síndrome de QT largo: distinguir las mutaciones patógenas de las variantes benignas. Circulation (2009) 120:1752-60. doi: 10.1161 / CIRCULATIONAHA.109.863076 Resumen de Pubmed / Texto Completo de Pubmed / Texto Cruzado Completo 28. Moss AJ, Shimizu W, Wilde AA, Towbin JA, Zareba W, Robinson JL, et al. Aspectos clínicos del síndrome de QT largo tipo 1 por ubicación, tipo de codificación y función biofísica de las mutaciones que afectan al gen KCNQ1. Circulation (2007) 115:2481-9. doi: 10.1161 / CIRCULATIONAHA.106.665406 Resumen de Pubmed / Texto Completo de Pubmed / Texto Cruzado Completo 29. Amin AS, Giudicessi JR, Tijsen AJ, Spanjaart AM, Reckman YJ, Klemens CA, et al. Las variantes en la región 3′ no traducida del canal de potasio KV7.1 codificado con KCNQ1 modifican la gravedad de la enfermedad en pacientes con síndrome de QT largo tipo 1 de una manera alélica específica. Eur Heart J (2012) 33: 714-23. doi: 10.1093/eurheartj / ehr473 Pubmed Resumen / Pubmed Texto Completo / Texto Cruzado 30. Barsheshet A, Goldenberg I, O-Uchi J, Moss AJ, Jons C, Shimizu W, et al. Mutaciones en los bucles citoplasmáticos del canal KCNQ1 y el riesgo de eventos potencialmente mortales: implicaciones para la respuesta específica de la mutación al tratamiento con bloqueadores β en el síndrome de QT largo tipo 1. Circulation (2012) 125:1988-96. doi: 10.1161 / CIRCULATIONAHA.111.048041 Resumen de Pubmed / Texto Completo de Pubmed / Texto Cruzado Completo 31. Schwartz PJ, Spazzolini C, Crotti L, Bathen J, Amlie JP, Timothy K, et al. The Jervell and Lange-Nielsen syndrome: natural history, molecular basis, and clinical outcome (en inglés). Circulation (2006) 113:783-90. doi: 10.1161 / CIRCULATIONAHA.105.592899 Resumen de Pubmed / Texto Completo de Pubmed / Texto Cruzado Completo 32. Bhuiyan AA, Wilde AA. IKs en el corazón y el oído, el oído puede hacer con menos que el corazón. Circ Cardiovasc Genet (2013) 6:141-3. doi: 10.1161 / CIRCGENÉTICA.113.000143 Texto completo cruzado 33. Wollnik B, Schroeder BC, Kubisch C, Esperer HD, Wieacker P, Jentsch TJ. Mecanismos fisiopatológicos de mutaciones dominantes y recesivas del canal K+ KVLQT1 encontradas en arritmias cardíacas hereditarias. Hum Mol Genet (1997) 6:1943-9. doi: 10.1093/hmg / 6.11.1943 Texto completo del texto cruzado 34. Wilde AA, Jongbloed RJ, Doevendans PA, Düren DR, Hauer RN, van Langen IM, et al. Los estímulos auditivos como desencadenantes de eventos arrítmicos diferencian a los pacientes relacionados con HERG (SQTL 2) de los pacientes relacionados con KVLQT1 (SQTL 1). J Am Coll Cardiol (1999) 33:327-32. doi: 10.1016 / S0735-1097(98)00578-6 Texto completo de CrossRef 35. Moss AJ, Zareba W, Kaufman ES, Gartman E, Peterson DR, Benhorin J, et al. Aumento del riesgo de eventos arrítmicos en el síndrome de QT largo con mutaciones en la región de los poros del canal de potasio del gen relacionado con el éter-a-go-go humano. Circulation (2002) 105:794-9. doi: 10.1161 | hc0702.105124 Resumen de Pubmed | Texto Completo de Pubmed / Texto Cruzado Completo 36. Lupoglazoff JM, Denjoy I, Villain E, Fressart V, Simon F, Bozio A, et al. Síndrome de QT largo en neonatos: trastornos de la conducción asociados con mutaciones en HERG y bradicardia sinusal con mutaciones en KCNQ1. J Am Coll Cardiol (2004) 43:826-30. doi: 10.1016 / j. jacc.2003.09.049 Resumen de Pubmed / Texto Completo de Pubmed / Texto Cruzado Completo 37. Chevalier P, Bellocq C, Millat G, Piqueras E, Potet F, Schott JJ, et al. Torsades de pointes complicando el bloqueo auriculoventricular: evidencia de una predisposición genética. Heart Rhythm (2007) 4:170-4. doi: 10.1016 / j. hrthm.2006.10.004 Resumen de Pubmed / Texto Completo de Pubmed / Texto Cruzado Completo 38. Hoorntje T, Alders M, van Tintelen P, van der Lip K, Sreeram N, van der Wal A, et al. Truncamiento prematuro homocigótico de la proteína HERG: el knockout HERG humano. Circulation (1999) 100:1264-7. doi: 10.1161 / 01.CIR.100.12.1264 Resumen de Pubmed / Texto Completo de Pubmed / Texto Cruzado Completo 39. Piippo K, Laitinen P, Swan H, Toivonen L, Viitasalo M, Pasternack M, et al. La homocigosis para una mutación del canal de potasio HERG causa una forma grave de síndrome de QT largo: la identificación de una mutación fundadora aparente en los finlandeses. J Am Coll Cardiol (2000) 35:1919-25. doi: 10.1016 / S0735-1097(00)00636-7 Resumen de Pubmed / Texto Completo de Pubmed / Texto Completo de CrossRef 40. Johnson WH Jr, Yang P, Yang T, Lau YR, Mostella BA, Wolff DJ, et al. Caracterización clínica, genética y biofísica de una mutación HERG homocigótica que causa síndrome de QT largo neonatal grave. Pediatr Res (2003) 53:744-8. doi: 10.1203 / 01.PDR.0000059750.17002.B6 Resumen de Pubmed / Texto Completo de Pubmed / Texto Cruzado Completo 41. Ackerman MJ, Tester DJ, Jones GS, Will ML, Burrow CR, Curran ME. Diferencias étnicas en las variantes del canal de potasio cardíaco: implicaciones para la susceptibilidad genética a la muerte súbita cardíaca y las pruebas genéticas para el síndrome congénito de QT largo. Mayo Clin Proc (2003) 78: 1479-87. doi:10.4065/78.12.1479 Resumen de Pubmed / Texto Completo de Pubmed / Texto Completo de CrossRef 42. Crotti L, Lundquist AL, Insolia R, Pedrazzini M, Ferrandi C, De Ferrari GM, et al. KCNH2-K897T es un modificador genético del síndrome de QT largo congénito latente. Circulation (2005) 112:1251-8. doi: 10.1161 / CIRCULATIONAHA.105.549071 Resumen de Pubmed / Texto Completo de Pubmed / Texto Cruzado Completo 43. Nof E, Cordeiro JM, Pérez GJ, Scornik FS, Calloe K, Love B, et al. Un polimorfismo de un solo nucleótido común puede exacerbar el síndrome de QT largo tipo 2 que conduce a la muerte súbita del lactante. Circ Cardiovasc Genet (2010) 3:199-206. doi: 10.1161 / CIRCGENÉTICA.109.898569 Resumen de Pubmed / Texto Completo de Pubmed / Texto Cruzado Completo 44. Bezzina C, Veldkamp MW, van Den Berg MP, Postma AV, Rook MB, Viersma JW, et al. Una única mutación en el canal Na(+) que causa síndromes de QT largo y de Brugada. Circ Res (1999) 85:1206-13. doi: 10.1161 / 01.RES.85.12.1206 Resumen de Pubmed / Texto Completo de Pubmed / Texto Completo de CrossRef 45. Kyndt F, Probst V, Potet F, Demolombe S, Chevallier JC, Baro I, et al. Nueva mutación de SCN5A que conduce a un defecto aislado de conducción cardíaca o al síndrome de Brugada en una gran familia francesa. Circulation (2001) 104:3081-6. doi: 10.1161 | hc5001.100834 Resumen de Pubmed | Texto Completo de Pubmed / Texto Cruzado Completo 46. Makita N, Behr E, Shimizu W, Horie M, Sunami A, Crotti L, et al. La mutación E1784K en el SCN5A se relaciona con el fenotipo clínico mixto del síndrome de QT largo tipo 3. J Clin Invest (2008) 118:2219-29. doi: 10.1172 / JCI34057 Resumen de Pubmed / Texto Completo de Pubmed / Texto Cruzado Completo 47. Keller DI, Acharfi S, Delacrétaz E, Benammar N, Rotter M, Pfammatter JP, et al. A novel mutation in SCN5A, delQKP 1507-1509, causing long QT syndrome: role of Q1507 residue in sodium channel inactivation. J Mol Cell Cardiol (2003) 35:1513-21. doi: 10.1016 / j. yjmcc.2003.08.007 Resumen de Pubmed / Texto Completo de Pubmed / Texto Cruzado Completo 48. Priori SG, Schwartz PJ, Napolitano C, Bloise R, Ronchetti E, Grillo M, et al. Estratificación de riesgo en el síndrome de QT largo. N Engl J Med (2003) 348:1866-74. doi: 10.1056 / NEJMoa022147 Resumen de Pubmed / Texto Completo de Pubmed / Texto Cruzado Completo 49. Lupoglazoff JM, Cheav T, Baroudi G, Berthet M, Denjoy I, Cauchemez B, et al. Mutación homocigótica en el SCN5A en el síndrome de QT largo con bloqueo auriculoventricular funcional de dos a uno. Circ Res (2001) 89:E16–21. doi: 10.1161 / hh1401. 095087 Texto completo del texto cruzado 50. Shi R, Zhang Y, Yang C, Huang C, Zhou X, Qiang H, et al. La mutación del canal cardíaco de sodio delQKP 1507-1509 se asocia con la expansión del espectro fenotípico del QT3, trastorno de conducción, miocardiopatía dilatada y alta incidencia de muerte súbita juvenil. Europace (2008) 10:1329-35. doi: 10.1093 / europace / eun202 Pubmed Resumen / Pubmed Texto Completo / Texto Cruzado 51. Zumhagen S, Veldkamp MW, Stallmeyer B, Baartscheer A, Eckardt L, Paul M, et al. Una mutación de deleción heterocigótica en el gen del canal de sodio cardíaco SCN5A con características de pérdida y ganancia de función se manifiesta como enfermedad de conducción aislada, sin signos de Brugada o síndrome de QT largo. PLoS One (2013) 8:e67963. doi: 10.1371 / journal.ponga.0067963 Resumen de Pubmed / Texto Completo de Pubmed / Texto Cruzado Completo 52. Planta LD, Bowers PN, Liu Q, Morgan T, Zhang T, State MW, et al. Una variante común del canal cardíaco de sodio asociada con la muerte súbita del lactante en afroamericanos, SCN5A S1103Y. J Clin Invest (2006) 116:430-5. doi: 10.1172 / JCI25618 Resumen de Pubmed / Texto Completo de Pubmed / Texto Cruzado Completo 53. Mohler PJ, Schott JJ, Gramolini AO, Dilly KW, Guatimosim S, duBell WH, et al. La mutación anquirina-B causa arritmia cardíaca de QT largo tipo 4 y muerte cardíaca súbita. Nature (2003) 421:634-9. doi: 10.1038 / nature01335 Resumen de Pubmed / Texto Completo de Pubmed / Texto Completo de CrossRef 54. Schott JJ, Charpentier F, Peltier S, Foley P, Drouin E, Bouhour JB, et al. Mapeo de un gen para el síndrome de QT largo con el cromosoma 4q25-27. Am J Hum Genet (1995) 57: 1114-22. Resumen de Pubmed | Texto Completo de Pubmed 55. Las proteínas Barhanin J, Lesage F, Guillemare E, Fink M, Lazdunski M, Romey G. K(V)LQT1 y lsK (Visón) se asocian para formar la corriente I(Ks) de potasio cardíaco. Nature (1996) 384:78-80. doi: 10.1038 / 384078a0 Resumen de Pubmed / Texto Completo de Pubmed / Texto Cruzado Completo 56. Sanguinetti MC, Curran ME, Zou A, Shen J, Spector PS, Atkinson DL, et al. Ensamblaje conjunto de proteínas K (V)LQT1 y Visón(IsK) para formar un canal de potasio I (Ks) cardíaco. Nature (1996) 384:80-3. doi: 10.1038 | 384080a0 Resumen de Pubmed / Texto Completo de Pubmed / Texto Cruzado Completo 57. Schulze-Bahr E, Wang Q, Wedekind H, Haverkamp W, Chen Q, Sun Y, et al. Las mutaciones en KCNE1 causan el síndrome de Jervell y Lange-Nielsen. Nat Genet (1997) 17:267-8. doi:10.1038/ng1197-267 Texto completo cruzado 58. Duggal P, Vesely MR, Wattanasirichaigoon D, Villafane J, Kaushik V, Beggs AH. Mutación del gen de IKs asociada con formas de síndrome de QT largo de Jervell y Lange-Nielsen y Romano-Ward. Circulation (1998) 97: 142-6. doi: 10.1186/1471-2350-9-24 Resumen de Pubmed / Texto Completo de Pubmed / Texto Completo de CrossRef 59. Nishio Y, Makiyama T, Itoh H, Sakaguchi T, Ohno S, Gong YZ, et al. D85N, un polimorfismo KCNE1, es una variante génica causante de enfermedad en el síndrome de QT largo. J Am Coll Cardiol (2009) 54:812-9. doi: 10.1016 / j. jacc.2009.06.005 Resumen de Pubmed / Texto Completo de Pubmed / Texto Cruzado Completo 60. Paulussen AD, Gilissen RA, Armstrong M, Doevendans PA, Verhasselt P, Smeets HJ, et al. Variaciones genéticas de KCNQ1, KCNH2, SCN5A, KCNE1 y KCNE2 en pacientes con síndrome de QT largo inducido por fármacos. Mol Med (2004) 82:182-8. doi: 10.1007 / s00109-003-0522-z Resumen de Pubmed / Texto Completo de Pubmed / Texto Cruzado Completo 61. Abbott GW, Sesti F, Splawski I, Buck ME, Lehmann MH, Timothy KW, et al. MiRP1 forma canales de potasio IKr con HERG y se asocia con arritmia cardíaca. Cell (1999) 97: 175-87. doi: 10.1016 / S0092-8674(00)80728-X Resumen de Pubmed / Texto Completo de Pubmed / Texto Cruzado Completo 62. Gordon E, Panaghie G, Deng L, Bee KJ, Roepke TK, Krogh-Madsen T, et al. Mutación de KCNE2 en un paciente con arritmia cardíaca inducida por estímulos auditivos y desequilibrio electrolítico sérico. Cardiovasc Res (2008) 77: 98-106. doi: 10.1093/cvr / cvm030 Resumen de Pubmed / Texto Completo de Pubmed / Texto Cruzado Completo 63. Yeso NM, Tawil R, Tristani-Firouzi M, Canún S, Bendahhou S, Tsunoda A, et al. Las mutaciones en Kir2. 1 causan los fenotipos eléctricos episódicos y de desarrollo del síndrome de Andersen. Cell (2001) 105:511-9. doi: 10.1016 / S0092-8674(01)00342-7 Resumen de Pubmed / Texto Completo de Pubmed / Texto Completo de CrossRef 64. Kubo Y, Baldwin TJ, Jan YN, Jan LY. Estructura primaria y expresión funcional de un canal de potasio rectificador interno de ratón. Nature (1993) 362: 127-33. doi: 10.1038 / 362127a0 Resumen de Pubmed / Texto Completo de Pubmed / Texto Cruzado Completo 65. Raab-Graham KF, Radeke CM, Vandenberg CA. Clonación molecular y expresión de un canal de potasio rectificador interno del corazón humano. Neuroreport (1994) 5:2501-5. doi: 10.1097/00001756-199412000-00024 Resumen de Pubmed / Texto Completo de Pubmed / Texto Completo de CrossRef 66. Splawski I, Timothy KW, Sharpe LM, Decher N, Kumar P, Bloise R, et al. Ca (V) 1.2 la disfunción de los canales de calcio causa un trastorno multisistémico que incluye arritmia y autismo. Cell (2004) 119:19-31. doi: 10.1016 / j.cell.2004.09.011 Resumen de Pubmed / Texto Completo de Pubmed / Texto Cruzado Completo 67. Splawski I, Timothy KW, Decher N, Kumar P, Sachse FB, Beggs AH, et al. Trastorno de arritmia grave causado por mutaciones cardíacas en los canales de calcio de tipo L. Proc Natl Acad Sci U S A (2005) 102:8089-96. doi:10.1073 / pnas.0502506102 Resumen de Pubmed / Texto Completo de Pubmed / Texto Cruzado Completo 68. Gillis J, Burashnikov E, Antzelevitch C, Blaser S, Gross G, Turner L, et al. QT largo, sindactilia, contracturas articulares, accidente cerebrovascular y nueva mutación CACNA1C: expandiendo el espectro del síndrome de Timothy. Am J Med Genet A (2012) 158A:182-7. doi: 10.1002 / ajmg.a.34355 Resumen de Pubmed / Texto Completo de Pubmed / Texto Cruzado Completo 69. Dufendach KA, Giudicessi JR, Boczek NJ, Ackerman MJ. El mosaicismo materno confunde el diagnóstico neonatal del síndrome de Timoteo tipo 1. Pediatría (2013) 131: e1991-5. doi: 10.1542 / pediatría.2012-2941 Resumen de Pubmed / Texto Completo de Pubmed / Texto Completo de CrossRef 70. Vatta M, Ackerman MJ, Ye B, Makielski JC, Ughanze EE, Taylor EW, et al. La caveolina-3 mutante induce corriente tardía persistente de sodio y se asocia con el síndrome de QT largo. Circulación (2006) 114:2104–12. doi: 10.1161 / CIRCULATIONAHA.106.635268 Resumen de Pubmed / Texto Completo de Pubmed / Texto Cruzado Completo 71. Cronk LB, Ye B, Kaku T, Tester DJ, Vatta M, Makielski JC, et al. Mecanismo novedoso para el síndrome de muerte súbita del lactante: corriente de sodio tardía persistente secundaria a mutaciones en caveolina-3. Heart Rhythm (2007) 4:161-6. doi: 10.1016 / j. hrthm.2006.11.030 Resumen de Pubmed / Texto Completo de Pubmed / Texto Cruzado Completo 72. Engelman JA, Zhang X, Galbiati F, Volonte D, Sotgia F, Pestell RG, et al. Molecular genetics of the caveolin gene family: implications for human cancers, diabetes, Alzheimer disease, and muscular dystrophy (en inglés). Am J Hum Genet (1998) 63: 1578-87. doi:10.1086/302172 Texto completo de CrossRef 73. Balijepalli RC, Foell JD, Hall DD, Hell JW, Kamp TJ. La localización de los canales cardíacos de tipo L Ca (2+) a un complejo de señalización macromolecular caveolar es necesaria para la regulación adrenérgica beta(2). Proc Natl Acad Sci U S A (2006) 103:7500-5. doi:10.1073 / pnas.0503465103 Resumen de Pubmed / Texto Completo de Pubmed / Texto Cruzado Completo 74. Medeiros-Domingo A, Kaku T, Tester DJ, Iturralde-Torres P, Itty A, Ye B, et al. Subunidad beta4 del canal de sodio codificado con SCN4B en el síndrome congénito de QT largo. Circulation (2007) 116:134-42. doi: 10.1161 / CIRCULATIONAHA.106.659086 Resumen de Pubmed / Texto Completo de Pubmed / Texto Cruzado Completo 75. Chen L, Marquardt ML, Tester DJ, Sampson KJ, Ackerman MJ, Kass RS. La mutación de una proteína ancladora de A-quinasa causa el síndrome de QT largo. Proc Natl Acad Sci U S A (2007) 104:20990-5. doi:10.1073 / pnas.0710527105 Resumen de Pubmed / Texto Completo de Pubmed / Texto Cruzado Completo 76. Wu G, Ai T, Kim JJ, Mohapatra B, Xi Y, Li Z, et al. Mutación alfa-1-sintrofina y Síndrome de QT largo: una enfermedad de alteración del canal de sodio. Circ Arrythm Electrophysiol (2008) 1:193-201. doi: 10.1161 / CIRCEP.108.769224 Resumen de Pubmed / Texto Completo de Pubmed / Texto Cruzado Completo 77. Yang Y, Yang Y, Liang B, Liu J, Li J, Grunnet M, et al. Identificación de un Kir3.4 mutación en el síndrome congénito de QT largo. Am J Hum Genet (2010) 86:872-80. doi: 10.1016 / j. ajhg.2010.04.017 Resumen de Pubmed / Texto Completo de Pubmed / Texto Cruzado Completo 78. Roden DM. Mecanismos y manejo de la proarritmia. Am J Cardiol (1998) 82:49I–57I. doi: 10.1016/S0002-9149(98)00472-X Texto completo cruzado 79. Mitcheson JS, Chen J, Lin M, Culberson C, Sanguinetti MC. Una base estructural para el síndrome de QT largo inducido por fármacos. Proc Natl Acad Sci U S A (2000) 97: 12329-33. doi:10.1073 / pnas.210244497 Texto completo del texto cruzado 80. Kannankeril PJ, Roden DM. QT largo inducido por fármacos y torsade de pointes: avances recientes. Curr Opin Cardiol (2007) 22:39-43. doi: 10.1097 / HCO.0b013e32801129eb Resumen de Pubmed / Texto Completo de Pubmed / Texto Cruzado Completo 81. Veerman CC, Verkerk AO, Blom MT, Klemens CA, Langendijk PN, van Ginneken AC, et al. El bloqueo de corriente de potasio del rectificador retardado lento contribuye de manera importante al síndrome de QT largo inducido por fármacos. Circ Arrythm Electrophysiol (2013) 6:1002-9. doi: 10.1161 / CIRCEP.113.000239 Resumen de Pubmed / Texto Completo de Pubmed / Texto Cruzado Completo 82. Sesti F, Abbott GW, Wei J, Murray KT, Saksena S, Schwartz PJ, et al. Polimorfismo común asociado con arritmia cardíaca inducida por antibióticos. Proc Natl Acad Sci U S A (2000) 97: 10613-8. doi:10.1073 / pnas.180223197 Resumen de Pubmed / Texto Completo de Pubmed / Texto Cruzado Completo 83. Kääb S, Crawford DC, Sinner MF, Behr ER, Kannankeril PJ, Wilde AA, et al. Un gran estudio de genes candidatos identifica el polimorfismo KCNE1 D85N como un posible modulador de torsades de pointes inducidos por fármacos. Circ Cardiovasc Genet (2012) 5:91-9. doi: 10.1161 / CIRCGENÉTICA.111.960930 Resumen de Pubmed / Texto Completo de Pubmed / Texto Cruzado Completo 84. Nakamura K, Katayama Y, Kusano KF, Haraoka K, Tani Y, Nagase S, et al. Síndrome de QT largo inducido por anticuerpos anti-KCNH2: nueva forma adquirida de síndrome de QT largo. J Am Coll Cardiol (2007) 50:1808-9. doi: 10.1016 / j. jacc.2007.07.037 Texto completo cruzado 85. Moss AJ, Zareba W, Benhorin J, Locati EH, Hall WJ, Robinson JL, et al. Patrones de onda T del ECG en formas genéticamente distintas del síndrome hereditario de QT largo. Circulation (1995) 92: 2929-34. doi: 10.1161 / 01.CIR.92.10.2929 Resumen de Pubmed / Texto Completo de Pubmed / Texto Cruzado Completo 86. Zhang L, Timothy KW, Vincent GM, Lehmann MH, Fox J, Giuli LC, et al. Espectro de patrones de onda ST-T y parámetros de repolarización en el síndrome congénito de QT largo: los hallazgos del ECG identifican genotipos. Circulation (2000) 102:2849-55. doi: 10.1161 / 01.CIR.102.23.2849 Resumen de Pubmed / Texto Completo de Pubmed / Texto Cruzado Completo 87. Tan HL, Bardai A, Shimizu W, Moss AJ, Schulze-Bahr E, Noda T, et al. Aparición de arritmias genotípicas específicas en el síndrome congénito de QT largo: posibles implicaciones terapéuticas. Circulation (2006) 114: 2096-103. doi: 10.1161 / CIRCULATIONAHA.106.642694 Resumen de Pubmed / Texto Completo de Pubmed / Texto Cruzado Completo 88. Locati EH, Zareba W, Moss AJ, Schwartz PJ, Vincent GM, Lehmann MH, et al. Diferencias relacionadas con la edad y el sexo en las manifestaciones clínicas en pacientes con síndrome congénito de QT largo: hallazgos del Registro Internacional de SQTL. Circulation (1998) 97: 2237-44. doi: 10.1161 / 01.CIR.97.22.2237 Resumen de Pubmed / Texto Completo de Pubmed / Texto Cruzado Completo 89. Zareba W, Moss AJ, Schwartz PJ, Vincent GM, Robinson JL, Priori SG, et al. Influencia del genotipo en el curso clínico del síndrome de QT largo. Grupo de Investigación del Registro Internacional de Síndrome de QT Largo. N Engl J Med (1998) 339:960-5. doi: 10.1056 / NEJM199810013391404 Pubmed Resumen / Pubmed Texto Completo / Texto Cruzado 90. Goldenberg I, Moss AJ, Bradley J, Polonsky S, Peterson DR, McNitt S, et al. Síndrome de QT largo después de los 40 años. Circulation (2008) 117: 2192-201. doi: 10.1161 / CIRCULATIONAHA.107.729368 Resumen de Pubmed / Texto Completo de Pubmed / Texto Cruzado Completo 91. Zareba W, Moss AJ, Locati EH, Lehmann MH, Peterson DR, Hall WJ, et al. Registro de Síndrome. Efectos moduladores de la edad y el sexo en el curso clínico del síndrome de QT largo por genotipo. J Am Coll Cardiol (2003) 42:103-9. doi: 10.1016 / S0735-1097(03)00554-0 Resumen de Pubmed / Texto Completo de Pubmed / Texto Completo de CrossRef 92. Seth R, Moss AJ, McNitt S, Zareba W, Andrews ML, Qi M, et al. Síndrome de QT largo y embarazo. J Am Coll Cardiol (2007) 49:1092-8. doi: 10.1016 / j. jacc.2006.09.054 Resumen de Pubmed / Texto Completo de Pubmed / Texto Cruzado Completo 93. Schwartz PJ, Priori SG, Dumaine R, Napolitano C, Antzelevitch C, Stramba-Badiale M, et al. Un vínculo molecular entre el síndrome de muerte súbita del lactante y el síndrome de QT largo. N Engl J Med (2000) 343:262-7. doi: 10.1056 / NEJM200007273430405 Texto completo del texto cruzado 94. Schwartz PJ, Priori SG, Bloise R, Napolitano C, Ronchetti E, Piccinini A, et al. Diagnóstico molecular en un niño con síndrome de muerte súbita del lactante. Lancet (2001) 358:1342-3. doi: 10.1016 / S0140-6736(01)06450-9 Resumen de Pubmed / Texto Completo de Pubmed / Texto Completo de CrossRef 95. Christiansen M, Tønder N, Larsen LA, Andersen PS, Simonsen H, Oyen N, et al. Mutations in the HERG K+-ion channel: a novel link between long QT syndrome and sudden infant death syndrome (en inglés). Am J Cardiol (2005) 95:433-4. doi: 10.1016 / j. amjcard.2004.09.054 Resumen de Pubmed / Texto Completo de Pubmed / Texto Cruzado Completo 96. Tester DJ, Ackerman MJ. Pruebas genéticas post mortem de síndrome de QT largo para detectar muerte súbita inexplicable en los jóvenes. J Am Coll Cardiol (2007) 49:240-6. doi: 10.1016 / j. jacc.2006.10.010 Resumen de Pubmed / Texto Completo de Pubmed / Texto Cruzado Completo 97. Hofman N, Tan HL, Clur SA, Alders M, van Langen IM, Wilde AA. Contribución de la cardiopatía hereditaria a la muerte cardíaca súbita en la infancia. Pediatrics (2007) 120:e967–73. doi: 10.1542 / pediatría.2006-3751 Resumen de Pubmed / Texto Completo de Pubmed / Texto Cruzado Completo 98. Wedekind H, Smits JP, Schulze-Bahr E, Arnold R, Veldkamp MW, Bajanowski T, et al. Mutación de novo en el gen SCN5A asociada con el inicio temprano de la muerte súbita del lactante. Circulation (2001) 104:1158-64. doi:10.1161/hc3501.095361 CrossRef Texto Completo 99. Wang DW, Desai RR, Crotti L, Arnestad M, Insolia R, Pedrazzini M, et al. Disfunción cardíaca del canal de sodio en el síndrome de muerte súbita del lactante. Circulation (2007) 115:368-76. doi: 10.1161 / CIRCULATIONAHA.106.646513 Resumen de Pubmed / Texto Completo de Pubmed / Texto Cruzado Completo 100. Van Norstrand DW, Valdivia CR, Tester DJ, Ueda K, London B, Makielski JC, et al. Molecular and functional characterization of novel glicerol-3-fosfato deshidrogenasa 1 like gen (GPD1-L) mutations in sudden infant death syndrome. Circulation (2007) 116:2253-9. doi: 10.1161 / CIRCULATIONAHA.107.704627 Resumen de Pubmed / Texto Completo de Pubmed / Texto Cruzado Completo 101. Tan BH, Pundi KN, Van Norstrand DW, Valdivia CR, Tester DJ, Medeiros-Domingo A, et al. Mutaciones asociadas al síndrome de muerte súbita del lactante en las subunidades beta del canal de sodio. Heart Rhythm (2010) 7: 771-8. doi: 10.1016 / j. hrthm.2010.01.032 Resumen de Pubmed / Texto Completo de Pubmed / Texto Cruzado Completo 102. Miller TE, Estrella E, Myerburg RJ, Garcia de Viera J, Moreno N, Rusconi P, et al. Pérdida fetal recurrente en el tercer trimestre y mosaicismo materno para el síndrome de QT largo. Circulation (2004) 109:3029-34. doi: 10.1161 / 01.CIR.0000130666.81539.9 E Pubmed Resumen / Pubmed Texto Completo / Texto Cruzado 103. Chockalingam P, Crotti L, Girardengo G, Johnson JN, Harris KM, van der Heijden JF, et al. No todos los betabloqueantes son iguales en el manejo del síndrome de QT largo tipos 1 y 2: recurrencias más altas de eventos bajo Metoprolol. J Am Coll Cardiol (2012) 60:2092-6. doi: 10.1016 / j. jacc.2012.07.046 Resumen de Pubmed / Texto Completo de Pubmed / Texto Cruzado Completo 104. Schwartz PJ, Priori SG, Cerrone M, Spazzolini C, Odero A, Napolitano C, et al. Denervación simpática cardíaca izquierda en el manejo de pacientes de alto riesgo afectados por el síndrome de QT largo. Circulation (2004) 109:1826-33. doi: 10.1161 / 01.CIR.0000125523.14403.1 E Pubmed Resumen / Pubmed Texto Completo / Texto Cruzado 105. Singh B, al Shahwan SA, Habbab MA, al Deeb SM, Biary N. Síndrome idiopático de QT largo: hacer la pregunta correcta. Lancet (1993) 341:741. doi:10.1016/0140-6736(93)90501-7 Texto completo de CrossRef 106. Gorgels AP, Al Fadley F, Zaman L, Kantoch MJ, Al Halees Z. El síndrome de QT largo con conducción auriculoventricular deteriorada: una variante maligna en bebés. J Cardiovasc Electrophysiol (1998) 9: 1225-32. doi: 10.1111 / j. 1540-8167. 1998.tb00096.x Resumen de Pubmed / Texto Completo de Pubmed / Texto Cruzado Completo 107. Kantoch MJ, Qurashi MM, Bulbul ZR, Gorgels AP. Un recién nacido con una cardiopatía congénita compleja, bloqueo auriculoventricular y taquicardia ventricular torsade de pointes. Pacing Clin Electrophysiol (1998) 21:2664-7. doi: 10.1111 / j. 1540-8159. 1998.tb00043.x Texto completo del texto cruzado 108. El-Hazmi MA, al-Swailem AR, Warsy AS, al-Swailem AM, Sulaimani R, al-Meshari AA. Consanguinidad entre la población de Arabia Saudita. J Med Genet (1995) 32:623-6. doi: 10.1136 / jmg.32.8.623 Texto completo cruzado 109. El Mouzan MI, Al Salloum AA, Al Herbish AS, Qurachi MM, Al Omar AA. Consanguinity and major genetic disorders in Saudi children: a community-based cross-sectional study. Ann Saudi Med (2008) 28:169-73. doi:10.4103/0256-4947.51726 Resumen de Pubmed / Texto Completo de Pubmed / Texto Completo de CrossRef 110. Medeiros-Domingo A, Bhuiyan DJ, Tester DJ, Hofman N, Bikker H, van Tintelen JP, et al. El receptor de rianodina codificado en RYR2/canal de liberación de calcio en pacientes diagnosticados previamente con taquicardia ventricular polimórfica catecolaminérgica o síndrome de QT largo inducido por el ejercicio con genotipo negativo: un análisis mutacional de marco de lectura abierto completo. J Am Coll Cardiol (2009) 54:2065-74. doi: 10.1016 / j. jacc.2009.08.022 Resumen de Pubmed / Texto Completo de Pubmed / Texto Cruzado Completo 111. Al-Aama JY, Al-Ghamdi S, Bdier AY, Wilde AA, BhuiyanA. Mutación de novo en el gen KCNQ1 causante del síndrome de Jervell y Lange-Nielsen. Clin Genet (2013). doi: 10.1111 / cge.12300 Resumen de Pubmed / Texto Completo de Pubmed / Texto Cruzado Completo 112. Splawski I, Timothy KW, Tateyama M, Clancy CE, Malhotra A, Beggs AH, et al. Variante del canal de sodio SCN5A implicada en el riesgo de arritmia cardíaca. Science (2002) 297:1333-6. doi: 10.1126 / ciencia.1073569 Texto completo del texto cruzado 113. Wilde AA, Bezzina CR. Genética de arritmias cardíacas. Heart (2005) 91: 1352-8.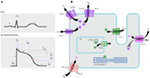
SQTL2
LQTS3
LQTS4
SQT5
LQTS6
LQTS7
SQT8
LQTS9 y LQTS10
LQTS11
LQTS12
LQTS13
SQTL adquirido
Las Características electrocardiográficas de las Tres Formas Comunes de Síndrome de QT Largo

Genotipo-Fenotipo
El manejo clínico del SQTL
SQTL: Perspectiva saudita
Declaración de Conflicto de Intereses
Agradecimientos