Ictiosis Congénita Autosómica Recesiva / Actas Dermo-Sifiliográficas
Introducción
La última clasificación consensuada de ictiosis diferencia entre 2 formas principales: las formas no sindrómicas, que se presentan con manifestaciones cutáneas solamente, y las formas sindrómicas, que también se presentan con manifestaciones en otros órganos (Tabla 1).1 Entre las formas no sindrómicas, se identifican 4 grupos: ictiosis comunes, ictiosis congénitas autosómicas recesivas (ARCIs), ictiosis queratinopáticas y otras ictiosis menos comunes.Tradicionalmente, el grupo de ARCIs se dividía en 2 trastornos, ictiosis lamelar (LI) y eritrodermia ictiosiforme congénita (CIE). En la nueva clasificación, la ictiosis arlequín (HI) se añadió a este grupo1 porque se han identificado mutaciones inactivadoras en el gen ABCA12 como responsables de este trastorno,2,3,mientras que las mutaciones sin sentido en el mismo gen pueden dar lugar al fenotipo LI4 o CIE5, 6. Otras variantes menos comunes incluidas en el grupo de ARCIs son el bebé colodión autocurativo (SHCB), acral SHCB e ictiosis de traje de baño.7-9
Clasificación Consensuada Basada en las Características Clínicas de la Ictiosis1.
| Nonddromic Formas | Sindrómico |
| Común IchthyosesIchthyosis vulgarisRecessive x-ligado a la ictiosis (no sindrómica )ajimajor formsHarlequin ichthyosisLamellar ichthyosisCongenital ichthyosiform erythrodermaMinor formsSelf-curación de colodión húmedo babyAcral de auto-sanación colodión húmedo babyBathing traje ichthyosisKeratinopathic IchthyosesMajor formsEpidermolytic Ichthyosissuperficial presentar queratosis ichthyosisminor formsannular presentar queratosis ichthyosiscurth-Macklin ichthyosisautosomal recesivo ictiosis epidermolítica Nevus epidermolítico Otras formassloricrina Queratodermaeritroqueratodermia vararabilis Sindrome de piel sensible ictiosiforme reticular congenital Eritroderm Síndrome de Maklick | Ictiosis ligada al cromosoma X recesiva Ictiosis folicular, alopecia y fotofobia (IFAP)Síndrome Síndrome de Scammann-happle (condrodisplasia punctata tipo 2) sindrómico de ictiosis autosómica trastornos de la piel sindrome de etherton sindrome de ciosis-hiptricosis sindrome de ciosis-Colangitis Esclerosantesdordenotricotistrofineurológicosjögren-Larsson syndromeRefsum diseaseMEDNIK syndromeFatal disease courseGaucher disease, type 2Multiple sulfatase deficiencyCEDNIK syndromeARC syndromeOther associated signsKID syndromeChanarin-Dorfman syndromeIchthyosis prematurity syndrome |
Abbreviations: ARC, arthrogryposis–renal dysfunction–cholestasis; ARCI, autosomal recessive congenital ichthyosis; CEDNIK, cerebral dysgenesis, neuropathy, ichthyosis, and palmoplantar keratoderma; KID, keratitis ichthyosis deafness; KLICK, keratosis linearis with ichthyosis congenital and sclerosing keratoderma; MEDNIK, retraso mental, enteropatía, sordera, neuropatía periférica, ictiosis, queratodermia.
Sólo se dispone de algunos datos sobre la epidemiología de la ARCIs. En los Estados Unidos, se ha estimado una prevalencia al nacer de 1 por cada 100 000 habitantes en el caso de las IF y de 1 por cada 200 000 habitantes en el caso de las CIE. Otros estudios han informado de una prevalencia combinada de LI y CIE de 1 por cada 200 000 a 300 000 habitantes.10,11 En algunos países, como Noruega, la prevalencia estimada es mayor (1 por 91 000) debido a mutaciones fundadoras.12 El hallazgo de 1 o varias mutaciones recurrentes en una población puede deberse a que la mutación se produjo en un momento dado de la historia y luego se pasó de generación en generación (mutación fundadora) o porque la región del genoma donde se encuentra la mutación tiene una secuencia de ADN susceptible a la mutación (punto caliente de mutación). En España, la prevalencia estimada de ARCI es de 1 por 138 000 en la población general y de 1 por 61 700 en niños menores de 10 años.13 En algunas regiones de España, la prevalencia podría ser aún mayor. En la costa gallega, por ejemplo, se reportó una prevalencia de 1 por 33 000, debido también a un efecto fundador.14
Ictiosis lamelar y Características eritrodermaclínicas Ictiosiformes Congénitas
Aunque originalmente se pensó que LI y CIE eran entidades diferentes, se han notificado casos de pacientes con manifestaciones clínicas intermedias y ambas afecciones pueden ser causadas por mutaciones en el mismo gen.15,16 Además, los pacientes con la misma mutación, incluso dentro de la misma familia, pueden desarrollar fenotipos diferentes.12,15
La mayoría de los pacientes nacen envueltos en una membrana de colodión que desaparece progresivamente durante las primeras semanas de vida y es reemplazada por el fenotipo definitivo (Fig. 1A). Hipohidrosis, intolerancia al calor severa y distrofia de uñas se observan con frecuencia tanto en LI como en CIE.17-19 Pacientes con LI suelen presentar manifestaciones clínicas más graves que los pacientes con EIC. Tienen grandes escamas en forma de placa, a menudo de color oscuro, que cubren toda la superficie corporal. El eritrodermia está ausente o es mínimo. Estos pacientes suelen tener ectropión y, a veces, eclabio, hipoplasia de cartílago articular y nasal, alopecia cicatrizante, especialmente en el borde del cuero cabelludo, y queratoderma palmoplantar (Fig. 1B y C). La EIC se caracteriza por la presencia de eritrodermia y escamas blanquecinas finas (Fig. 2). Algunos pacientes presentan eritema marcado y descamación generalizada. Las escamas pueden ser grandes y de color oscuro, particularmente en las superficies extensoras de las patas. En los casos menos graves, el eritema es leve y la descamación está bien.

Características clínicas de ictiosis lamelar. A, Descamación laminar pardusca. B, Hiperqueratosis plantar marcada. C, Alopecia cicatrizante del cuero cabelludo.
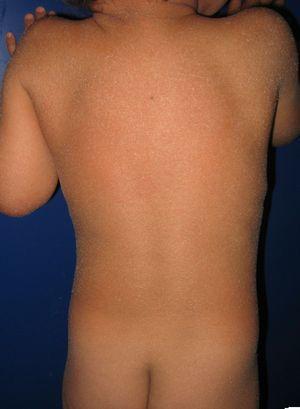
Paciente con eritrodermia ictiosiforme congénita y mutaciones en el gen ALOXE3. Se puede observar eritema leve y descamación pelurácea blanquecina generalizada.
la Histopatología
cambios Histopatológicos no proporcionan un diagnóstico. En LI, se observa hiperqueratosis ortoqueratótica masiva, generalmente con el doble de extensión que en CIE. La epidermis es acantótica y ocasionalmente adquiere un aspecto similar a la psoriasis. La tasa de proliferación celular es normal o ligeramente elevada.17-19 Pacientes con EIC tienen hiperqueratosis menos marcada, con paraqueratosis focal o extensa, una capa granular normal o engrosada y acantosis más pronunciada. El recambio epidérmico aumenta.17-19
Ultraestructura
Aunque hasta ahora no se ha encontrado una estrecha correlación entre los hallazgos moleculares, clínicos y ultraestructurales, la microscopía electrónica puede ser útil para descartar otras formas de ictiosis y para guiar los análisis genéticos en algunos casos. Se han descrito cuatro tipos de ictiosis congénita (Tabla 2).
Clasificación Ultraestructural de Ictiosis Congénitas.
| Tipo | Característica principal | Otras características | Mutaciones | Manifestaciones clínicas |
| 1 | Ausencia de marcadores ultraestructurales de ictiosis tipos 2, 3 y 4 | Gotas lipídicas o anillos en el estrato córneo (más frecuente)Pequeños granulados de queratohialina gránulos de revestimiento de membrana lobular o vascular | TGM1 (33.3%)ALOX12B (2 casos) | CIE |
| 2 | el Colesterol hendiduras en el estrato córneo | Ausencia o adelgazamiento de la córnea envelopeSmall keratohyalin granulesLipid gotitas | TGM1 (89-100%) | LI |
| 3 | Laminado membraneous estructuras en el estrato granuloso y/o estrato córneo. | Granulados anormales de revestimiento de membrana Gotitas lipídicas Asociadas a vacuolas yuxtanucleares prominentes en la capa granular | NIPAL4 (93%) | CIE (más frecuente)LI |
| 4 | Paquetes de membrana trilamelar que llenan algunas células en el estrato granuloso y / o estrato córneo | gránulos de recubrimiento de membrana anormales | FTAP4 | Ictiosis síndrome de prematuridad (100%) |
Abreviaturas: CIE, eritrodermia ictiosiforme congénita; LI, ictiosis lamelar.
Ictiosis congénita Tipo 1
La ictiosis congénita tipo 1 se caracteriza por la ausencia de marcadores ultraestructurales para ictiosis tipos 2, 3 y 4. Por lo tanto, el diagnóstico generalmente solo se realiza cuando se han excluido los otros tipos. El hallazgo más frecuente es la presencia de gotas lipídicas o anillos en el estrato córneo (Fig. 3A).20 Estas gotas lipídicas no son una característica constante o específica de este tipo en particular,ya que no están presentes en todos los casos, 20 y pueden estar presentes en otros tipos de ictiosis.21,22 Clínicamente, la mayoría de los pacientes presentan manifestaciones de EIC.12,20 Un tercio de los pacientes tienen mutaciones en el gen TGM1.16 Este tipo ultraestructural también se ha identificado en asociación con mutaciones en el gen ALOX12B.23,24

Imágenes de microscopio electrónico. A, Ictiosis congénita tipo 1, que muestra gotitas lipídicas en el estrato córneo y ausencia de marcadores ultraestructurales de los otros tipos de ictiosis. B, Ictiosis congénita tipo 2, caracterizada por la presencia de hendiduras de colesterol (flecha) en los corneocitos.
Ictiosis congénita Tipo 2
La ictiosis congénita tipo 2 se caracteriza por hendiduras de colesterol en el estrato córneo (Fig. 3B).21 Tales hendiduras son un hallazgo constante en este tipo de ictiosis, y se pueden detectar en diferentes biopsias en el mismo paciente; el tratamiento con retinoides orales no tiene impacto en estas hendiduras.También se han observado 12,25 agregados densos de electrones en los corneocitos en algunos pacientes con actividad deficiente de la TGasa 1.26-28 Clínicamente, la mayoría de los pacientes presentan manifestaciones graves de EIC.12 Este tipo ultraestructural está fuertemente asociado con mutaciones en el gen TGM1.12,16
Ictiosis congénita Tipo 3
La ictiosis congénita tipo 3 se caracteriza por estructuras membranosas lamelares en el estrato granuloso y/o el estrato córneo. Estas estructuras están dispuestas en tiras alrededor de un espacio vacío cerca del núcleo.22,29 – 31 Las manifestaciones clínicas en este tipo son diferentes a las demás; la aparición de ictiosis es variable, la descamación y el eritema pueden ser irregulares o generalizados, y las flexiones en particular se ven afectadas. Las mutaciones en el gen NIPAL4 son responsables del 93% de las ictiosis tipo 3.32
Ictiosis congénita Tipo 4
Característicamente, en la ictiosis congénita tipo 4, algunas células en el estrato granuloso y el estrato córneo están llenas de paquetes de membrana trilamelar.33 Estos hallazgos son patognómicos para ictiosis síndrome de prematuridad, una afección actualmente considerada como una forma sindrómica de ictiosis.34,35
Estudios moleculares
En términos genéticos, los ARCIs son muy heterogéneos. El gen TGM1 se relaciona con la mayoría de los casos, pero se han notificado mutaciones en otros 5 genes (ALOX12B, ALOXE3, NIPAL4, CYP4F22 y ABCA12). Fischer et al.36 estudiaron a 520 familias con ARCI e identificaron mutaciones en al menos 1 de estos genes en 78% de los casos (TGM1 en 32%, NIPAL4 en 16%, ALOX12B en 12%, CYP4F22 en 8%, ALOXE3 en 5% y ABCA12 en 5%). En otro estudio de 250 pacientes con ARCI de diferentes orígenes, 38% tenían mutaciones en TGM1, 6,8% tenían mutaciones en ALOXE3 y 6,8% tenían mutaciones en ALOX12B.37 En Galicia, se identificaron mutaciones en los genes TGM1, ALOX12B, ALOXE3, NIPAL4 y CYP4F22 en el 75% de las familias estudiadas, pero la distribución de las mutaciones fue diferente.14 El gen TGM1 fue mutado en 68.el 7% de los casos, mientras que el gen ALOXE3 estaba mutado en solo 1 paciente. No detectamos mutaciones en ninguno de los otros 3 genes estudiados.
TGM1
El gen TGM1 se encuentra en el cromosoma 14q11.2 y tiene 15 exones (GenBank NM-000359.2). Codifica la enzima TGasa 1, que es una de las 3 enzimas TGasa que se encuentran en la epidermis.38 Esta enzima participa en la formación de la envoltura cornificada catalizando la reticulación dependiente del calcio de varias proteínas, como involucrina, loricrina y proteínas ricas en prolina.39,40 también cataliza la unión de ??- hidroxiceramidas en la capa externa de la envoltura cornificada con proteínas en la capa interna.41,42 En pacientes con mutaciones en TGM1, falta la envoltura cornificada y la actividad de la TGasa 1 es reducida o inexistente.43-47
Desde 1995, cuando se identificó este gen como responsable de algunos casos de ARCI, 48-50 se han notificado más de 110 mutaciones en pacientes de orígenes diferentes. Las mutaciones en TGM1 son la causa más común de ARCI.36,37 Esta mutación se ha encontrado en el 55% de los casos en los Estados Unidos y en el 84% de los casos en Noruega.12,51 La mutación más frecuente es c.877-2A> G, que se ha encontrado en el 34% de los alelos mutados reportados hasta la fecha.52 La alta frecuencia de esta mutación en países como Estados Unidos y Noruega se debe a un efecto fundador.12,53 La segunda mutación más frecuente es la p.Arg142His. Esta y otras mutaciones similares se han notificado en países como Egipto, Alemania, Finlandia y los Estados Unidos15, 49-51,54-56,y parece que se trata de mutaciones hotspot.57 La mutación p. Arg307Trp es frecuente en la población japonesa.5 En Galicia, la p. Arg760X, c. 1223_1227delACACA y c.Se identificaron 984 + 1G>A mutaciones en TGM1 en el 81,82% de las familias con mutaciones en este gen, lo que sugiere un efecto fundador.14 La confirmación de esta hipótesis se obtuvo mediante un estudio de haplotipos (trabajo aún no publicado).
Las mutaciones en TGM1 son responsables de la mayoría de los casos de LI15,27,44,46,56,58-63 y para un pequeño porcentaje de casos de CIE.43,47,64,65 Tales mutaciones también pueden dar lugar a otras formas de ARCI como SHCB, acral SHCB e ictiosis de traje de baño.
Muchos estudios han intentado demostrar asociaciones genotipo-fenotipo entre mutaciones en TGM1 y hallazgos ultraestructurales o clínicos, pero hasta la fecha no se ha observado una correlación significativa.15,16,53 En general, los pacientes con mutaciones en el gen TGM1 se ven más gravemente afectados que aquellos sin tales mutaciones. En un estudio de 83 pacientes con ARCI en Suecia y Estonia, la presencia de ectropión y colodión bebé se asoció con mutaciones en TGM1, mientras que se observó una mayor tasa de eritema en pacientes sin mutaciones en este gen.66 Otro estudio mostró que el tipo de descamación es la principal diferencia entre portadores y no portadores de mutaciones en TGM1, al encontrar que todos los pacientes con mutaciones en este gen tenían descamación lamelar, mientras que el 80% de los que no tenían mutaciones en TGM1 tenían descamación fina.14 Además, se ha visto que las mutaciones truncantes se asocian con mayor frecuencia con hipohidrosis y trastornos de la sudoración que las mutaciones sin sentido.51 En la población de América del Norte, un modelo basado en la presencia de ciertas características clínicas predice que los pacientes que nacen en colodión y tienen trastornos oculares o alopecia tienen 4 veces más probabilidades de tener mutaciones en TGM1.51
ALOXE3 y ALOX12B
Los genes ALOXE3 y ALOX12B están ubicados en el cromosoma 17p13.1. 67 Tienen una estructura similar con 15 exones que codifican los LOX epidérmicos eLOX-3 y 12R-LOX.68,69 El hecho de que se expresen predominantemente en las capas suprabasales de la epidermis apoya su papel en las fases avanzadas de diferenciación epidérmica, con participación en el procesamiento de cuerpos laminares.24,70 Estas enzimas actúan en pasos adyacentes en la vía de la hepoxilina (Fig. 4). 12R-LOX transforma el ácido araquidónico en ácido 12R-hidroxieicosatetraenoico, mientras que eLOX-3 convierte este producto en un isómero de epoxialcohol69,71 de la familia de la hepoxilina A3.72 El producto de hepoxilina es inestable y se hidroliza en células a un derivado trihidroxi específico (trioxilina). Aunque no se conoce el papel exacto de los productos de la vía de la hepoxilina, se ha especulado que pueden participar en la formación de lípidos intercelulares del estrato córneo o actuar como señales para inducir la diferenciación de queratinocitos.
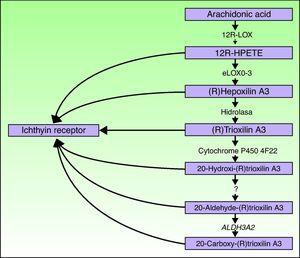
Diagrama esquemático de la vía de la hepoxilina, que muestra la participación de los genes ALOXE3, ALOX12B, NIPAL4 y CYP4F22. Las mutaciones en estos genes son responsables de algunos tipos de ARCI. HPETE indica ácido hidroperoxieicosatetraenoico.
Los genes ALOX12B y ALOXE3 se identificaron por primera vez en 200273,74.Desde entonces, se han notificado más de 30 mutaciones en el gen ALOX12B23,24,37,75-77 y aproximadamente 10 en el gen ALOXE337,74,75. Estas mutaciones son responsables del 14% al 17% de ARCIs36,37 y 72.2% de SHCBs.23,78,79 La relación causal entre estas mutaciones y el fenotipo se confirmó demostrando que la actividad catalítica del LOX epidérmico se abolió totalmente en pacientes con estas mutaciones75,80 y utilizando modelos animales que reprodujeron el fenotipo ictiosiforme observado en humanos.81-83 Ambos genes son responsables de un porcentaje similar de casos de ARCI. Sin embargo, el rango de diferentes mutaciones en el gen ALOXE3 es limitado, debido al predominio de 2 mutaciones, p. Arg234X y p. Pro630Leu, que parecen corresponder a puntos calientes.37,74,75
Los pacientes con mutaciones en los genes ALOXE3 y ALOX12B suelen mostrar un fenotipo CIE.74,75,77 La gravedad de la descamación es leve o moderada, y las escamas tienen un color blanquecino o marrón claro. También puede haber eritema. Hasta el 76% de los pacientes nacen como bebés de colodión y el 88% tienen trastornos de sudoración.37 Pacientes con mutaciones en el gen ALOX12B muestran una descamación blanquecina más limitada en comparación con los portadores de mutaciones en el gen ALOXE3. En estos casos, las escamas son de color marrón y adherentes. La presencia de eritema, hiperqueratosis palmoplantar y acentuación de los pliegues palmoplantar también se asocian con mutaciones en ALOX12B.37
Ichthyin/NIPAL4
El gen NIPAL4, también conocido como el gen icthyin, se encuentra en el cromosoma 5q33. Tiene 6 exones que codifican una proteína con varios dominios transmembrana de función desconocida.84 Se ha planteado la hipótesis de que el producto proteico participa en la misma vía metabólica que el LOX y puede actuar como receptor de trioxilinas A3 y B3 o de otros metabolitos de la vía metabólica de hepoxilina.84 Por lo tanto, estaría implicado en la formación de cuerpos laminares o en su transporte hacia el espacio extracelular.32 En apoyo de esto hay 2 observaciones. En primer lugar, en el 93% de los casos, las mutaciones en este gen se asocian con un patrón ultraestructural de ictiosis congénita tipo 3, caracterizado por anomalías en los cuerpos lamelares y la presencia de membranas perinucleares alargadas en el estrato granuloso.32 En segundo lugar, NIPAL4 se expresa esencialmente en el estrato granuloso de la epidermis, donde están presentes los cuerpos laminares.85
Desde el descubrimiento del gen NIPAL4 en 200484, solo se han notificado 9 mutaciones en pacientes de países mediterráneos (Argelia, Turquía y Siria),84 países escandinavos,32 Pakistán,85 Islas Feroe,32 y América del Sur.84
El espectro clínico de pacientes con mutaciones en este gen es amplio, incluso entre miembros de la misma familia. Entre el 3,7% 32 y el 60% 84 nacen en colodión. Cuando la membrana del colodión desaparece, la mayoría de los pacientes desarrollan las manifestaciones de EIC, con escamas blanquecinas finas en una base eritematosa en la cara y el tronco y escamas marrones más grandes en el cuello, las nalgas y las piernas.Puede haber xerosis marcada, placas hiperqueratóticas reticulares parduzcas generalizadas que aparecen acentuadas en los pliegues de la piel y discromia facial.32,85 Además, la queratoderma palmoplantar es un hallazgo frecuente junto con contracturas ocasionales de los dedos y uñas curvas. Algunos estudios han reportado hallazgos más típicos de la LI.32,85 Se ha notificado la presencia de signos y síntomas de dermatitis atópica en algunos pacientes, aunque no se detectaron mutaciones en el gen FLG en ninguno de estos casos.85
CYP4F22
El gen FLJ39501 o CYP4F22 se encuentra en el cromosoma 19p13.12. 86 Tiene 12 exones87 y codifica un citocromo P450, familia 4, subfamilia F, polipéptido 2, homólogo de la leucotrieno B4 – ω-hidroxilasa (CYP4F2). La reacción catalizada por el producto de FLJ39501 en la piel y los sustratos de dicha reacción puede deducirse por analogía con sus homólogos conocidos CYP4F2 y CYP4F3.88 Se ha planteado la hipótesis de que CYP4F2 y CYP4F3 participan en la vía de la hepoxilina al catalizar la conversión de trioxilina A3 a 20-hidroxi-(R)trioxilina A387 y que el producto final de esta vía, 20-carboxi-trioxilina A3, puede tener un efecto regulador biológico clave en la piel.89
Hasta la fecha, solo se han notificado 8 mutaciones de este gen en 12 familias consanguíneas de países mediterráneos87 y en 1 familia de origen israelí.62
En las familias reportadas por Lefèvre et al., 87 la mayoría de los pacientes tenían un fenotipo CIE al nacer, que posteriormente evolucionó a LI, y los pacientes solían nacer con eritrodermia marcada, aunque sin membrana de colodión. A medida que envejecían, desarrollaban una descamación generalizada de color gris blanquecino, que era más marcada en la región periumbilical, en las nalgas y en la parte inferior del cuerpo. La hiperlinealidad de las palmas y plantas de los pies y la descamación en el cuero cabelludo, a veces de tipo pitiriasiforme, fueron frecuentes.87 En otra familia, los 3 miembros afectados nacieron como bebés colisionados y desarrollaron eritrodermia intensa, descamación generalizada y queratoderma palmoplantar.62
ABCA12
En 2003, se informó que el gen ABCA12 era responsable de algunos casos de LI y se mapeó al cromosoma 2q34.4 Posteriormente se confirmó que las mutaciones en este gen también eran responsables de la HI.2, 3ABCA12 codifica 53 exones y pertenece a una familia de transportadores ABC, que se unen al trifosfato de adenosina al tiempo que facilitan el transporte de varias moléculas a través de la membrana celular.90 Los miembros de la subfamilia ABCA están implicados en el transporte de lípidos.La función deficiente de ABCA12 causa trastornos de transporte de lípidos en los cuerpos lamelares y, por lo tanto, conduce a una disminución de los niveles de lípidos intercelulares en el estrato córneo.3Estudios ultraestructurales han demostrado que ABCA12 se encuentra en cuerpos laminares asociados con glicosilceramidas.Las mutaciones de 91ABCA12 se han asociado con trastornos en la distribución y transporte de glicosilceramidas y con niveles disminuidos de hidroxiceramidas, uno de los principales componentes de la barrera lipídica en los espacios intercelulares.3,6,92,93 La hiperqueratosis masiva que ocurre en estos pacientes podría ser una respuesta compensatoria a una barrera lipídica deficiente.94 También podría deberse a la falta de descamación de los corneocitos,93 que podría ser causada por defectos en el transporte de ciertas proteasas, como la calicreina 5 y la catepsina D, resultantes de trastornos en los cuerpos laminares.95 Modelos murinos y estudios in vitro sugieren que las mutaciones ABCA12 también tienen un efecto en la diferenciación epidérmica.95-97
Hasta la fecha, se han notificado más de 50 mutaciones en el gen ABCA12 en pacientes con ARCI de África, Europa, Pakistán y Japón. Las mutaciones más frecuentes son p. Val244SerfsTer28,2, 98,99 identificadas en poblaciones pakistaníes e indias, y p. Asn1380Ser, 4 identificadas en familias africanas. En ambos casos, estas pueden ser mutaciones fundacionales.
La extensión de las mutaciones ABCA12 está relacionada con el fenotipo, con mutaciones asociadas con la pérdida completa de la función que conduce al fenotipo HI.2,3,98 – 102 Por el contrario, en LI y CIE, la mayoría de las mutaciones son sin sentido y tienen un efecto menos grave en la función de las proteínas.4-6, 103 Las mutaciones subyacentes al fenotipo LI parecen estar concentradas en la primera región de casete de unión de trifosfato de adenosina.4 Clínicamente, los pacientes con CIE y mutaciones en el gen ABCA12 tienen escalas de tamaño mediano que son algo más grandes que las que generalmente se observan en pacientes con este fenotipo.
Ictiosis de arlequín
HI o feto de arlequín es una forma grave y generalmente fatal de ictiosis. Los niños suelen ser prematuros con extensas placas hiperqueratóticas brillantes, separadas por profundas fisuras, que cubren todo el tegumento y forman patrones geométricos que recuerdan a la ropa usada por los arlequines, dando así nombre a la condición. La tirantez de la piel conduce a una marcada eversión de los párpados y los labios, al desarrollo rudimentario del cartílago articular y nasal y, ocasionalmente, a la microcefalia. Los niños rara vez tienen pestañas o cejas, aunque el cabello en el cuero cabelludo puede conservarse. Las manos y los pies están hinchados y edematosos, y a menudo cubiertos por una capa similar a un guante. Pueden tener contracturas en los dedos.
Para estos pacientes, el riesgo de morir durante el período neonatal es muy alto.104 La ventilación pulmonar está comprometida; la pérdida transepidérmica de agua conduce a deshidratación, desequilibrio hidroeléctrico e inestabilidad térmica; y el riesgo de infecciones aumenta. La opresión facial y el eclabio dificultan la succión y, por lo tanto, la alimentación, con el correspondiente empeoramiento de la deshidratación. Los neonatos con esta afección rara vez vivieron más de unas pocas semanas. En los últimos años, sin embargo, las posibilidades de supervivencia a largo plazo han aumentado notablemente, debido principalmente a la administración de retinoides sistémicos y al progreso en la atención neonatal intensiva.105 En un estudio reciente, el 83% de los pacientes tratados con retinoides orales sobrevivieron en comparación con el 24% de los pacientes no tratados. La mayoría de las muertes ocurrieron en los primeros 3 días de vida, pero el tratamiento no se inició hasta después de esto en muchos de los sobrevivientes.104 Esto sugiere que muchas de estas muertes tempranas habrían ocurrido independientemente del tratamiento con retinoides.
Los niños que sobreviven al período neonatal generalmente desarrollan EIC grave.La naturaleza y ubicación de las mutaciones en el gen ABCA12 y el grado de pérdida de la función del transportador pueden determinar el pronóstico.3,92,107 Pacientes que conservan un cierto grado de actividad proteica, aunque mínima, pueden tener una mejor probabilidad de sobrevivir. Los portadores de mutaciones homocigotas tienen una tasa de mortalidad más alta.104
La principal característica histológica del HI es la presencia de un estrato córneo ortoqueratótico extremadamente grueso y compacto. Los folículos pilosos y los conductos sudoríparos tienen tapones hiperqueratóticos107,108 prominentes y tienen cuerpos lamelares anormales o ausentes, inclusiones de lípidos o restos de orgánulos o núcleos en los corneocitos, y ausencia de lípidos intercelulares en el estudio ultraestructural.108.109 Los folículos pilosos muestran una marcada concentración de material queratótico, que es una característica diagnóstica de la HI utilizada para el diagnóstico prenatal.
Hasta la fecha, la tasa de detección de mutaciones en el gen ABCA12 en pacientes con HI es cercana al 100%, por lo que parece ser una condición genéticamente homogénea.
Bebé en Colodión y Bebé en Colodión autocurativo
Los bebés en Colodión suelen nacer prematuramente y la morbilidad y mortalidad perinatales aumentan. Al nacer, el neonato está cubierto por una membrana transparente brillante que recuerda a la envoltura de celofán (Fig. 5). Los bebés tienen ectropión, eclabio e hipoplasia del cartílago nasal y articular. La succión y la ventilación pulmonar puede ser hindered110 y la pérdida transepidérmica de agua y el riesgo de las infecciones es mayor.110,111

Bebé colodión que posteriormente progresó a un fenotipo de ictiosis lamelar.
Colodión húmedo bebé es la presentación habitual de HI y CIE. La LI autosómica dominante112, 113 el síndrome de Sjögren-Larsson,110 la tricotiodistrofia,114 la enfermedad de Gaucher juvenil, 110 la enfermedad de almacenamiento de lípidos neutros, el síndrome de Conradi-Hünermann-Happle, el síndrome de Hays-Wells y la displasia ectodérmica 115 también pueden presentarse ocasionalmente como colodión bebé. La membrana desaparece espontáneamente en el 10% al 24% de los neonatos, para dar paso a una piel completamente normal.110.116 En el pasado, estos casos se describían como LI del recién nacido,117 pero no se conocen como SHCB.118 Algunos autores han sugerido el término ictiosis colodionaria auto-mejorada porque muchos de estos pacientes, cuando se reexaminan más tarde en la infancia o en la adultez, tienen un grado variable de anhidrosis e intolerancia al calor y signos leves de ictiosis, como xerosis y descamación fina, particularmente en las axilas y el cuello.78
Ni la microscopía óptica ni las investigaciones ultraestructurales del bebé colodión son específicas. Por lo tanto, es preferible retrasar la biopsia de piel hasta que se haya desarrollado el fenotipo definitivo.
Se han identificado mutaciones en los genes TGM1,7,119ALOXE3,78 y ALOX12B23,78,79 en pacientes con SHCB. Las mutaciones de ALOX12B son las más comunes. En una serie de 15 pacientes escandinavos con SHCB, el 67% tenía mutaciones en el gen ALOX12B, el 25% en el gen ALOXE3 y el 8,3% en el gen TGM1.No se encontraron 78 mutaciones en algunos pacientes, por lo que es probable que otros genes también estén implicados. Se ha especulado que estas mutaciones reducen la actividad enzimática en el útero, pero no después del nacimiento.7 En el útero, donde la presión hidrostática es alta, la quelación por agua convierte la enzima mutada en una conformación inactiva. Después del nacimiento, cuando la presión disminuye, la enzima vuelve a su forma activa y su actividad aumenta lo suficiente para mantener un fenotipo normal o mínimamente afectado.7
Bebé Colodión autocurativo Acral
Aunque el bebé colodión afecta a todo el cuerpo, se han notificado casos limitados a las regiones acrales. En 1952, Finlay et al.120 reportaron un caso de membrana de colodión que afectó solo las manos y los pies y que siguió un curso de autocuración. Recientemente, se ha notificado un nuevo caso de acral SHCB en asociación con mutaciones del gen TGM1.8 No se sabe por qué estas lesiones están restringidas a las regiones acrales, aunque pueden estar en funcionamiento factores asociados con la regulación de la actividad enzimática dependiente del sitio.8
Ictiosis en traje de baño
La ictiosis en traje de baño se notificó por primera vez como una variante independiente de ARCI en 2005, aunque anteriormente se habían notificado casos de ictiosis con una distribución peculiar.121-123 Se ha detectado principalmente en pacientes de origen sudafricano,9 aunque también se ha reportado en los individuos de Europa y países Mediterráneos.124 Al nacer, los pacientes tienen una membrana de colodión generalizada que luego se desprende para dejar la distribución característica de la descamación. El tronco, la región proximal de los brazos, incluidas las axilas, el cuello y el cuero cabelludo generalmente se ven afectados, mientras que la parte central de la cara, las extremidades y la región suprarrenal generalmente se salvan.9 Las escamas son grandes, laminares y de color oscuro. La descamación más fina puede ocurrir en las fosas poplíteas y antecubitales.124,125 Las palmas de las manos y las plantas de los pies tienen hiperqueratosis difusa leve, mientras que la parte posterior de las manos y los pies no muestran compromiso.
El estudio histopatológico de la piel afectada muestra hiperqueratosis marcada sin paraqueratosis, capas granulares normales, acantosis leve o moderada y un infiltrado linfocítico leve en la dermis superior.9 Las observaciones de microscopía electrónica son consistentes con ictiosis congénita tipo 2 en la mayoría de los casos. La piel no involucrada no muestra ningún hallazgo anormal.124,125 En la piel sana, la actividad de la TGasa 1 se reduce ligeramente y generalmente se localiza en áreas pericelulares. En la piel afectada, la actividad enzimática es residual y se encuentra anormalmente en el citoplasma.Se han detectado 124 mutaciones
en el gen TGM1 en todos los pacientes con ictiosis en traje de baño estudiados hasta la fecha.119,124 – 126 La mutación más común es p. Arg315Leu, que se ha identificado en la mayoría de los pacientes sudafricanos y podría ser una mutación fundadora. Oji et al.124 sugería que la temperatura de la piel podría desempeñar un papel en el desarrollo de estas manifestaciones. Utilizando termografía digital, los autores mostraron una fuerte correlación entre la temperatura corporal y la descamación, siendo las áreas más calientes del cuerpo las más afectadas. Aufenvenne et al.127 mostraron una disminución de la temperatura óptima para la actividad de la TGasa 1 en pacientes con ictiosis en traje de baño. Esta disminución no se observó en controles sanos ni en pacientes con LI generalizada, lo que explicaría el fenotipo de estos pacientes. La temperatura óptima es de 37°C para la enzima normal y de 31°C para la enzima mutada.
Tratamiento
El objetivo principal del tratamiento en ictiosis es eliminar la descamación y reducir la xerosis sin causar irritación excesiva (Tabla 3). Antes de decidir el tratamiento, se deben tener en cuenta aspectos como la edad y el sexo del paciente, el tipo y la gravedad de la enfermedad, y la extensión y el lugar de las lesiones.128
Estrategia Terapéutica en Ictiosis Congénitas Autosómicas Recesivas.
| Estrategia terapéutica para las ictiosis congénitas autosómicas recesivas | |
| Baño y eliminación mecánica de escamas | Baño con bicarbonato de sodio o almidón de trigo, almidón de maíz o almidón de arroz; extracción mecánica de las básculas (1 o 2 veces al día) |
| Tratamiento tópico (secuencial) | Humectantes con urea Queratinolíticos combinados con propilEnglicol queratinolíticos combinados (propilenglicol, α-hidroxi ácidos o urea)Queratinolíticos combinados con ácido salicílico Retinoides tópicos En neonatos y niños pequeños, aplicar un vehículo sin ingredientes activos. Evitar la urea, el ácido salicílico y el ácido láctico debido al riesgo de absorción sistémica |
| Tratamiento oral | Retinoides orales (acitretina o isotretinoína) |
| Otras medidas | Seguimiento del ectropión por el oftalmólogo Limpieza regular del oído externo por el especialista en oído, garganta y nariz Fisioterapia para prevenir contracturas.Evitar actividades extenuantes en una alta temperatura ambientehidroterapia |
Baño y Eliminación Mecánica de Escamas
Se recomienda el baño diario para los pacientes con ARCI para eliminar mecánicamente escamas y restos de crema hidratante. Esto es más fácil si el paciente se sumerge en agua durante 15 a 30 minutos. Algunos autores recomiendan agregar bicarbonato de sodio al baño para desnaturalizar las queratinas y hacer que el agua sea alcalina, y así facilitar la eliminación de las escamas.129 Otros productos que se pueden agregar incluyen almidón de trigo, almidón de maíz o almidón de arroz. Los aceites de baño no son apropiados, ya que pueden provocar oclusión con el consiguiente riesgo de proliferación bacteriana y empeoramiento de la termorregulación.
Tratamiento tópico
Las cremas hidratantes y los agentes queratolíticos tópicos suelen ser la primera opción terapéutica. Mejoran la función de barrera de la piel y facilitan la descamación. Pueden producirse efectos adversos locales leves, como prurito transitorio, irritación o sensación de escozor.
El cloruro de sodio, la urea, el acetato de vitamina E, el glicerol y la vaselina se pueden usar como humectantes y lubricantes. En pacientes con descamación gruesa e hiperqueratosis marcada, se pueden añadir 1 o más agentes queratolíticos,como α-hidroxi ácidos (ácido láctico y glicólico), 130 ácido salicílico,N-acetilcisteína,131-133 urea (>5%), 134 y propilenglicol. También se utilizan moduladores de diferenciación de queratinocitos. Estos incluyen retinoides tópicos (tretinoína, adapaleno, tazaroteno),135,136 calcipotriol,137 y dexpantenol.Los retinoides tópicos a menudo causan irritación y fisuras pequeñas y muy dolorosas.137 Además, existe un riesgo de absorción y teratogenicidad en mujeres fértiles si se utilizan demasiado.138 Para mejorar la eficacia de los queratolíticos y los humectantes, se puede aplicar apósito oclusivo en áreas específicas refractarias al tratamiento.139 También se puede lograr un efecto aditivo o sinérgico combinando 2 o más agentes queratolíticos o humectantes.140-142 El tratamiento debe optimizarse para cada individuo, dada la naturaleza altamente variable de la afección y la sensibilidad de la piel y las diferencias en respuesta a cada tratamiento. El proceso de optimización se puede ayudar tratando un lado del cuerpo de manera diferente al otro para permitir comparaciones. Los recién nacidos y los niños pequeños deben tratarse con un vehículo sin sustancias activas, ya que la piel es muy fina y sensible y la mayoría de los queratolíticos no se toleran. Además, el riesgo de absorción percutánea de productos tópicos como la urea, el ácido salicílico y el ácido láctico es mayor.143-145
Tratamiento sistémico
Los retinoides orales tienen efectos queratolíticos que ayudan a eliminar las escamas y prevenir la hiperqueratosis excesiva. Tanto la isotretinoína como los retinoides aromáticos (acitretina y etretinato) han demostrado ser eficaces en el tratamiento de ARCIs.128,146,147 Acitretina en una dosis de 0,5 a 1 mg/kg/día es el fármaco más utilizado, especialmente en pacientes con LI.148 Los pacientes con CIE pueden tener una respuesta más completa y en dosis más bajas.
Los principales efectos adversos son trastornos mucocutáneos, teratogenicidad, trastornos musculoesqueléticos y perfil lipídico anormal y elevación de transaminasas.149-152 Con respecto a la teratogenicidad, en el caso del etretinato y la acitretina, se deben evitar los medicamentos durante el embarazo y las pacientes deben evitar quedar embarazadas durante 3 años después de la interrupción del tratamiento.151 La isotretinoína tiene una vida media más corta y se elimina completamente del organismo después de 1 mes, por lo que puede ser la opción preferida en mujeres que desean quedarse embarazadas.128
La monitorización del tratamiento debe incluir un análisis de laboratorio con una prueba de función hepática y un perfil lipídico antes de iniciar el tratamiento, a continuación, 1 mes y 3 meses después de iniciar el tratamiento. En mujeres en edad fértil, se debe realizar una prueba de embarazo en las 2 semanas anteriores al inicio del tratamiento y se debe utilizar un método anticonceptivo eficaz desde las 4 semanas anteriores al tratamiento hasta los 3 años posteriores (en el caso de acitretina). Cuando se requiera un tratamiento prolongado con retinoides, se debe controlar el crecimiento y el desarrollo óseo. Algunos autores sugieren realizar un estudio óseo antes del tratamiento seguido de un examen anual.151 Las guías recientes no recomiendan realizar una radiografía de rutina debido a los posibles efectos dañinos.152 En cambio, se recomiendan estudios radiográficos selectivos en pacientes con dolor óseo atípico.152
Una alternativa al tratamiento con retinoides sistémicos es el uso de fármacos conocidos como agentes bloqueadores del metabolismo del ácido retinoico, que aumentan los niveles endógenos de ácido retinoico. Uno de estos medicamentos es el liarozol, al que la Agencia Europea de Medicamentos y la Administración de Alimentos y Medicamentos de los Estados Unidos le han concedido el estatus de huérfano para el tratamiento de LI, CIE e HI.153-155 Se ha demostrado que este medicamento es más eficaz que la acitretina en ensayos clínicos y también se tolera mejor y tiene un mejor perfil farmacocinético.154
Otros cuidados médicos
En pacientes con ectropión, la aplicación de lágrimas artificiales y lubricantes para los ojos e hidratación de la piel de la cara y las mejillas en particular puede reducir la retracción palpebral. La corrección quirúrgica es una opción válida en casos graves, pero por lo general debe repetirse unos años después. La hidroterapia puede ser beneficiosa.Se debe aconsejar a los pacientes que eviten la actividad física intensa cuando la temperatura ambiente sea alta, dado que la hipohidrosis conlleva el riesgo de insolación y convulsiones. Los retinoides orales pueden mejorar la termorregulación.157 La fisioterapia es importante para prevenir la contractura de flexión, particularmente en el caso de la HI. La limpieza regular del conducto auditivo externo por un especialista en oído, garganta y nariz puede evitar que se acumulen escamas y, por lo tanto, evitar la pérdida de audición.
Asesoramiento Genético y Diagnóstico Prenatal
Cuando se diagnostica ictiosis a un paciente, se le debe ofrecer asesoramiento genético apropiado en el que se explique la naturaleza del trastorno, el modo de transmisión y el riesgo de manifestaciones futuras en la familia. El diagnóstico prenatal puede indicar si el feto está afectado y, si este es el caso, se puede ofrecer preparación psicológica de la familia y anticipar problemas durante el embarazo y el parto. A los padres se les puede dar la opción de un aborto si no hay tratamiento disponible. Además, si la terapia génica para estas afecciones estuviera disponible en el futuro, el diagnóstico prenatal permitiría la aplicación de esta terapia lo antes posible.
Durante más de 20 años, el diagnóstico prenatal se realizó tomando una muestra de biopsia de piel fetal y estudiándola por microscopía óptica, microscopía electrónica o inmunohistoquímica.158,159 Este procedimiento invasivo solo se pudo realizar en las últimas fases del embarazo, entre las semanas 15 y 23 de gestación, y se asoció con un riesgo de 1 a 3% de perder el feto.160.161 La identificación de los mecanismos moleculares de los trastornos hereditarios de la piel ha permitido un diagnóstico mucho más temprano basado en técnicas genéticas.102,162-164 El ADN fetal se obtiene por amniocentesis realizada entre las semanas 15 y 20 o por muestreo de vellosidades coriónicas entre las semanas 10 y 12. El riesgo de pérdida fetal con estas técnicas es menor de entre 0,5% y 1%.165 Otros métodos no invasivos en desarrollo son el análisis de ADN de células fetales y ADN fetal libre en circulación maternal166, así como el uso de ultrasonido tridimensional.167,168
El diagnóstico genético preimplantacional también podría ser posible en técnicas de fertilización in vitro, de modo que solo se implanten en el útero óvulos fertilizados libres de la mutación, evitando así la necesidad de aborto en la mayoría de los casos.169
Estrategias futuras para el Tratamiento Genético de la Ictiosis
Aunque se han logrado avances importantes en el diagnóstico genético de la ictiosis, también se están aplicando nuevas estrategias para estas enfermedades.170 La piel es el órgano más accesible para las terapias de transferencia de genes, por lo que estas técnicas son mínimamente invasivas.171 Sin embargo, la piel también tiene características inmunológicas únicas que no favorecen la expresión a largo plazo de un producto transgénico.En LI, un proceso de transferencia génica ex vivo logró restaurar la expresión normal de TGM1 y corregir el fenotipo de piel trasplantada en la parte posterior de ratones inmunosuprimidos.173.174 Recientemente, también se ha recuperado el fenotipo de queratinocitos cultivados de pacientes con HI debido a mutaciones en el gen ABCA12.3
Conflictos de intereses
Los autores declaran no tener conflictos de intereses.