La genética del labio leporino y paladar hendido
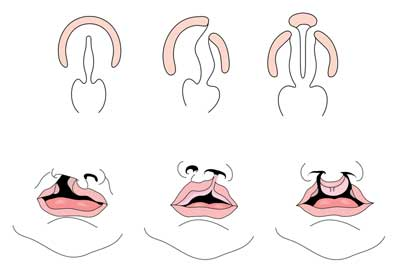 Intro/abstractel labio izquierdo con o sin paladar hendido es una anomalía congénita compleja que puede aislarse o verse junto con otras malformaciones. También puede ser parte del fenotipo de un síndrome genético. Este artículo sirve como una revisión de la prevalencia de labio leporino y paladar hendido, los riesgos de recurrencia y los riesgos de otras anomalías congénitas. Se explorarán síndromes genéticos y exposiciones teratogénicas que se sabe están asociadas con hendiduras orales. Además, se discutirán las pruebas genéticas comúnmente solicitadas en el entorno genético clínico pediátrico para la evaluación del paciente con labio leporino y paladar hendido.
Intro/abstractel labio izquierdo con o sin paladar hendido es una anomalía congénita compleja que puede aislarse o verse junto con otras malformaciones. También puede ser parte del fenotipo de un síndrome genético. Este artículo sirve como una revisión de la prevalencia de labio leporino y paladar hendido, los riesgos de recurrencia y los riesgos de otras anomalías congénitas. Se explorarán síndromes genéticos y exposiciones teratogénicas que se sabe están asociadas con hendiduras orales. Además, se discutirán las pruebas genéticas comúnmente solicitadas en el entorno genético clínico pediátrico para la evaluación del paciente con labio leporino y paladar hendido.
Labio leporino con o sin paladar hendido (CL/PC) difiere de un paladar hendido aislado (PC) en niveles embrionarios, epidemiológicos y genéticos. El labio leporino suele ser el resultado de que la prominencia maxilar y la prominencia nasal medial no se fusionan entre la quinta y la sexta semana de desarrollo embrionario. Normal desarrollo de paladar resultados de la formación del paladar primario y secundario paladar. El paladar primario se forma en las semanas seis a siete por el desarrollo y fusión de los procesos nasal medial, nasal lateral y maxilar. El paladar secundario se origina en los estantes palatales (que se desarrollan a partir de los procesos maxilares pares del primer arco branquial) convirtiéndose en horizontal y fusionándose, formando los paladares duros y blandos alrededor de la novena semana de desarrollo embrionario. Los estantes también se fusionan con el paladar primario y el tabique nasal. (1)
Las hendiduras orales son uno de los defectos de nacimiento más comunes observados en el vivero neonatal, con una prevalencia general de 1.6 por mil recién nacidos en todo el mundo, con CL/PC visto en aproximadamente uno por mil nacimientos y PC visto en 0,6 por mil nacimientos. (2) Hay una mayor frecuencia de CL/PC en individuos de ascendencia asiática, africana y nativa americana. El CL / PC también es más común en los hombres. En cambio, no hay diferencias significativas en la incidencia de PC entre los diferentes orígenes étnicos, y la PC es más común en las mujeres. (3) Los riesgos de recurrencia dentro de una familia dependen de si la hendidura está aislada (sin otros hallazgos clínicos presentes) o se ve como parte de un síndrome genético. La mayoría de los casos de hendiduras orales están aislados (aproximadamente el 80%). Se cree que las hendiduras aisladas tienen herencia multifactorial: se deben a una combinación de múltiples factores, tanto genéticos como ambientales. El riesgo de recurrencia (Tabla 1) aumenta cuando hay más de un familiar afectado. El riesgo de recurrencia también aumenta cuanto más grave sea el defecto.

El labio leporino y el paladar hendido se pueden ver con otras anomalías congénitas. La probabilidad de una etiología genética o teratogénica aumenta las anomalías más congénitas que presenta un paciente. La presencia de otros problemas como discapacidad intelectual, problemas de comportamiento como autismo, características dismórficas u otras preocupaciones médicas también hará que sea más probable un trastorno genético o una exposición teratogénica. Aproximadamente el 13% de las personas con labio leporino tendrán otros problemas médicos o anomalías. El número aumenta al 37% con labio leporino y paladar hendido y al 47% con paladar hendido solo.
Se ha demostrado que la exposición prenatal a agentes teratogénicos (como talidomida, anticonvulsivos, alcohol, ácido retinoico y cigarrillos) y enfermedades maternas (como diabetes, rubéola y deficiencia de folato) aumenta el riesgo de hendiduras orales. La presencia de bandas amnióticas también aumenta el riesgo de hendiduras. Se sabe que la suplementación periconceptual con ácido fólico reduce el riesgo de hendiduras orales.
La secuencia de Pierre Robin es una anomalía craneofacial caracterizada por hipoplasia o micrognacia mandibular, paladar hendido secundario en forma de U y glosoptosis que conduce a apnea obstructiva y dificultades de alimentación. La secuencia de Pierre Robin puede verse como parte de síndromes genéticos (síndrome de deleción 22q11.2, síndrome de Stickler; descrito a continuación). (5)
Hay cientos de síndromes genéticos asociados con hendiduras orales, incluidas anomalías citogenéticas (aneuploidías, microdeleciones) y trastornos de un solo gen (mendelianos). La confirmación de un diagnóstico genético es esencial para determinar el pronóstico y establecer un riesgo de recurrencia.
Las aneuploidías como la trisomía 13 y 18 tienen una fuerte asociación con el CL/PC. La trisomía 13 (también conocida como síndrome de Patau) se asocia con tres copias del cromosoma 13, o translocaciones Robertsonianas desequilibradas que involucran al cromosoma 13. Los bebés que nacen con esta afección generalmente mueren en el período neonatal. Las características clínicas incluyen labio leporino y paladar hendido, retraso del crecimiento, malformaciones nerviosas centrales graves (incluida la holoprosencefalia), microcefalia, microptalmia, coloboma del iris, ausencia de ojos, orejas malformadas, polidactilia, puños apretados, pies basculantes, defectos cardíacos congénitos y defectos urogenitales. En la trisomía 13 se pueden observar hendiduras de la línea media (por lo demás muy raras) debido al riesgo de defectos de la línea media, incluida la holoprosencefalia. La trisomía 18 (también conocida como síndrome de Edwards) se debe típicamente a tres copias distintas del cromosoma 18, y se asocia con un desenlace postnatal deficiente. Las características clínicas incluyen labio leporino y paladar hendido, discapacidad intelectual, retraso en el crecimiento, cardiopatía congénita, hipertonía, micrognatia, esternón corto, orejas malformadas de conjunto bajo, manos apretadas, pies basculantes y uñas hipoplásicas, entre otras. La trisomía 13 y 18 se puede confirmar o descartar fácilmente haciendo análisis cromosómicos (cariotipado).
Los síndromes de microdeleción generalmente implican la eliminación de parte de un cromosoma. Estas eliminaciones pueden ser demasiado pequeñas para ser detectadas mediante cariotipado estándar y pueden requerir la detección de FISH (hibridación fluorescente in situ) o tecnología de microarray. Un síndrome de microdeleción bien conocido asociado con el paladar hendido es el síndrome de deleción 22q11.2 (también conocido como síndrome Digeorge/Velocardiofacial). Las anomalías palatales que incluyen incompetencia velofaríngea, hendiduras submucosas, úvula bífida y paladar hendido se observan en 69% de los individuos con deleción 22q11.2 y pueden formar parte de la secuencia de Pierre Robin. Otros hallazgos clínicos incluyen enfermedades cardíacas congénitas, pérdida de audición, características dismórficas, deficiencia inmunitaria, hipocalcemia, anomalías renales, problemas de alimentación, anomalías esqueléticas y trastornos psiquiátricos. Se cree que aproximadamente el 10% de los casos de síndrome de deleción 22q11.2 son familiares. La deleción se segrega de manera autosómica dominante.(6) El síndrome de Wolf-Hirschhorn, que se debe a una deleción en el brazo corto del cromosoma 4, también está asociado con hendiduras orales (en el 25% al 50% de los individuos afectados). Los rasgos faciales característicos (incluyendo glabelas prominentes que conducen a la “apariencia de casco de guerrero griego”), enfermedades cardíacas congénitas, discapacidad intelectual, convulsiones, retraso en el crecimiento, micrognatia, marcas o hoyos preauriculares e hipodoncia también se pueden ver como parte de la afección.(7)
Los trastornos de un solo gen con hendiduras orales incluyen el síndrome de Stickler, el síndrome de Treacher Collins y el síndrome de Van der Woude, entre muchos otros. El síndrome de Stickler es un trastorno del colágeno con herencia autosómica dominante y, menos comúnmente, autosómica recesiva. Las características comunes incluyen paladar hendido (visto como parte de la secuencia de Pierre Robin o sin micrognatia), pérdida de audición (neurosensorial y conductiva), hallazgos esqueléticos (artritis de inicio temprano, displasia espondiloepifisaria), anomalías oculares (miopía alta, anomalías vítreas) y rasgos faciales característicos (con subdesarrollo del maxilar y el puente nasal, retrusión de la cara media). Las pruebas genéticas para el síndrome de Stickler pueden ser complejas, ya que se han descrito mutaciones en al menos seis genes en individuos afectados. Aproximadamente 90% de los pacientes con síndrome de Stickler tienen mutaciones en el gen COL2A1 y tienen una forma autosómica dominante de la afección.(8) El síndrome de Treacher Collins es una afección autosómica dominante caracterizada por paladar hendido con o sin labio leporino en el 28% de los individuos afectados. Otras anomalías incluyen hipoplasia de los huesos cigomáticos y la mandíbula, anomalías del oído externo, coloboma del párpado inferior, pérdida auditiva conductiva, ausencia de pestañas inferiores, desplazamiento del vello preauricular en las mejillas y estenosis coanal o atresia. El diagnóstico del síndrome de Treacher Collins se basa en hallazgos clínicos y radiográficos. Se han descrito mutaciones en al menos tres genes, con mutaciones en TCOF1 observadas en 78 a 93% de los pacientes.(9) El síndrome de Van der Woude se caracteriza por la presencia de fístulas congénitas, generalmente bilaterales, paramedianas del labio inferior (fosas), o a veces pequeños montículos con un tracto sinusal que sale de una glándula mucosa del labio, y hendiduras orales (incluyendo CL/CP y CP). Van der Woude es una afección autosómica dominante asociada con mutaciones en el gen IRF6 (10). Las pruebas para afecciones de un solo gen o de varios genes requieren un análisis directo del gen mediante secuenciación y/o análisis de deleción / duplicación (como MLPA).
Dado que los síndromes genéticos con labio leporino y paladar hendido pueden estar asociados con aneuploidías, microdeleciones/microduplicaciones cromosómicas o trastornos de un solo gen, las pruebas genéticas pueden ser un proceso complicado. Un historial médico completo, un pedigrí de tres generaciones, un historial de embarazo y un examen dismorfológico realizado por un genetista clínico pueden aclarar el cuadro clínico y permitir pruebas genéticas dirigidas. Las tecnologías más nuevas, como el microarray, permitirán la identificación de pequeñas microdeleciones y microduplicaciones que antes no se realizaban en el cariotipado estándar. Desafortunadamente, esta técnica también conduce a la identificación de eliminaciones y duplicaciones de significado clínico desconocido, lo que complica el proceso de asesoramiento genético. Las pruebas para detectar trastornos de un solo gen o trastornos mendelianos requieren la disponibilidad clínica de pruebas genéticas para el gen deseado. También puede ser costoso si no está cubierto por un seguro médico. Las nuevas tecnologías, como la secuenciación de última generación, la secuenciación del exoma o la secuenciación del genoma (conocidas colectivamente como pruebas genómicas), ahora están disponibles clínicamente. Al analizar de cientos a miles de genes simultáneamente, estas pruebas aumentan significativamente la potencia y el rendimiento diagnósticos. En comparación con otras técnicas, estas pruebas pueden proporcionar una respuesta más rápida y de una manera más rentable. En el campo de la investigación, la secuenciación de exomas y genomas ha llevado a la identificación de nuevos genes, así como a la expansión de las características clínicas y el espectro de mutaciones genéticas. Al igual que con la tecnología de microarrays, las pruebas genómicas pueden detectar síndromes que no están relacionados con la presentación del paciente y/o el motivo de la prueba. Dada la complejidad inherente de las pruebas genéticas, es necesario el consentimiento informado.
Conclusión
Aunque el labio leporino y el paladar hendido son una anomalía aislada en la mayoría de los casos, existe una fuerte asociación entre las hendiduras orales y otras anomalías y síndromes genéticos. Una evaluación genética por parte de un genetista clínico y un asesor genético es esencial para la orientación anticipada y para determinar los riesgos de recurrencia. Las pruebas genéticas, que requieren el consentimiento informado, se pueden coordinar e interpretar durante una evaluación genética.
Anya Revah, MS, es consejera sénior en genética en la División de Genética Médica del Hospital Maimónides de Bebés y Niños en Brooklyn, Nueva York. También es miembro activo del Equipo Multidisciplinario de Labio Leporino y Paladar Hendido del Centro Médico Maimónides y del Hospital del Condado de Kings. Tiene una Maestría en Ciencias en Asesoramiento Genético de la Universidad de Boston en Boston, Massachusetts.
1. Sadler TW. Embriología Médica de Langman. Novena Edición. Páginas 390-395.
2. Parker SE, Mai CT, Canfield MA, Rickard R, Wang Y, Meyer RE, Anderson P, Mason CA, Collins JS, Kirby RS, Correa A. Para la Red Nacional de Prevención de Defectos de Nacimiento. Estimaciones nacionales actualizadas de prevalencia de nacimientos para defectos de nacimiento seleccionados en los Estados Unidos. 2004-2006. Investigación de defectos de nacimiento (Parte A): Teratología Clínica y Molecular 2010;88: 1008-1016.
3. Fraser FC. La genética del labio leporino y el paladar hendido. Ser. J. Hum. Genet. 1970;22: 336–352.
4. Van Rooij IA, Ocke MC, et al. La ingesta periconceptual de folato mediante suplementos y alimentos reduce el riesgo de labio leporino no sindrómico con o sin paladar hendido. Prev Med 2004; 39: 689-694.
5. Tan TY. Kilpatrick N, Farlie PG. Perspectivas genéticas y de desarrollo de la secuencia de Pierre Robin. Ser. J. Med. Genet. 2013; 163C: 295-305.
6. McDonald-McGinn DM, Emanuel BS, Zackai EH. síndrome de deleción 22q11.2. Sept. 23, 1999. . En: Pagon RA, Adam MP, Ardinger HH, et al., Editor. GeneReviews . Seattle (WA): Universidad de Washington, Seattle; 1993-2014. Disponible desde: http://www.ncbi.nlm.nih.gov/books/NBK1523/.
7. Battaglia A, Carey JC, South ST, et al. Síndrome de Wolf-Hirschhorn. Abr. 29, 2002. . En: Pagon RA, Adam MP, Ardinger HH, et al., Editor. GeneReviews . Seattle (WA): Universidad de Washington, Seattle; 1993-2014. Disponible en: http://www.ncbi.nlm.nih.gov/books/NBK1183/.
8. Robin NH, Moran RT, Síndrome de Ala-Kokko L. Stickler. Jun. 9, 2000. . En: Pagon RA, Adam MP, Ardinger HH, et al., Editor. GeneReviews . Seattle (WA): Universidad de Washington, Seattle; 1993-2014. Disponible desde: http://www.ncbi.nlm.nih.gov/books/NBK1302/.
9. Katsanis SH, Jabs EW. Síndrome de Treacher Collins. Jul. 20, 2004. . En: Pagon RA, Adam MP, Ardinger HH, et al., Editor. GeneReviews . Seattle (WA): Universidad de Washington, Seattle; 1993-2014. Disponible a partir de: http://www.ncbi.nlm.nih.gov/books/NBK1532/.
10. Schutte BC, Saal HM, Goudy S, et al. Trastornos Relacionados con IRF6. Oct. 30, 2003. . En: Pagon RA, Adam MP, Ardinger HH, et al., Editor. GeneReviews . Seattle (WA): Universidad de Washington, Seattle; 1993-2014. Disponible desde: http://www.ncbi.nlm.nih.gov/books/NBK1407/.