Mucopolisacaridosis: signos de diagnóstico temprano en bebés y niños
Las mucopolisacaridosis (MPS) son un grupo de enfermedades clínicamente heterogéneas causadas por deficiencias de las enzimas lisosomales necesarias para la descomposición de los glucosaminoglicanos (GAG).
Las MPS se caracterizan por manifestaciones clínicas progresivas y sistémicas. A pesar de su heterogeneidad bioquímica y genética, los diferentes tipos comparten características clínicas clave en combinaciones variables, incluyendo displasia articular y esquelética con rigidez (excepto MPS IV donde hay laxitud) y dolor, rasgos faciales gruesos, opacidad corneal, hernias inguinales o abdominales, infecciones recurrentes del tracto respiratorio superior, enfermedad de las válvulas cardíacas, síndrome del túnel carpiano y afectación neurológica variable. Estas características generalmente aparecen en los primeros meses de vida para las formas severas y en la primera infancia para las formas más atenuadas, pero a menudo se subestiman y generalmente se tienen en cuenta solo cuando son claramente evidentes.
La rareza de estos trastornos y la variabilidad en la presentación clínica con frecuencia conducen a un retraso diagnóstico; esto puede variar de meses, para las formas graves, a años, para las atenuadas (ver Rigoldi et al. en este suplemento ). Sin embargo, debido a la rápida progresión y la necesidad urgente de intervención en las presentaciones severas, incluso meses de retraso en el diagnóstico pueden producir resultados catastróficos para la salud futura del paciente.
Nuestro objetivo es subrayar en esta revisión los signos tempranos de las formas graves que deben alertar al pediatra en su primera aparición.
¿Qué signos / síntomas podemos esperar en niños con formas graves de MPS?
Los síntomas y signos que sugieren diferentes formas de MPS se notifican en la Tabla 1 según la edad. En los primeros 6 meses de vida, se pueden observar hernias inguinales, infecciones respiratorias anormalmente frecuentes, otitis y organomegalia en pacientes con MPS, particularmente en MPS I (síndrome de Hurler), que tiene el inicio más precoz y grave. Además, de 6 a 12 meses, estos bebés muy a menudo desarrollan un gibus (cifosis toraco-lumbar), soplo cardíaco, hernia umbilical, hipotonía leve y retraso en el crecimiento . También pueden tener la típica apariencia facial gruesa (Fig. 1). Las MPS II, VI y VII pueden presentar un fenotipo temprano similar. Los pacientes con MPS IV tienen un fenotipo esquelético temprano con la aparición de displasia de cadera en los primeros meses de vida y tórax de paloma (pectus carinatum) en los primeros 12-18 meses. Los bebés MPS II presentan un compromiso similar, pero de aparición posterior. En el segundo año de vida, los bebés con síndrome de Hurler MPS I desarrollan retraso cognitivo, mientras que un paciente con síndrome de Hurler MPS II puede ser identificado solo debido a un crecimiento excesivo, infecciones frecuentes de las vías respiratorias y múltiples cirugías ; los rasgos faciales típicos aparecen solo más tarde. Algunos pacientes con MPS II también desarrollan hiperactividad entre los 18 y los 24 meses. En el tercer año de vida, la hiperactividad y el retraso cognitivo son fácilmente reconocibles en MPS II y MPS III.
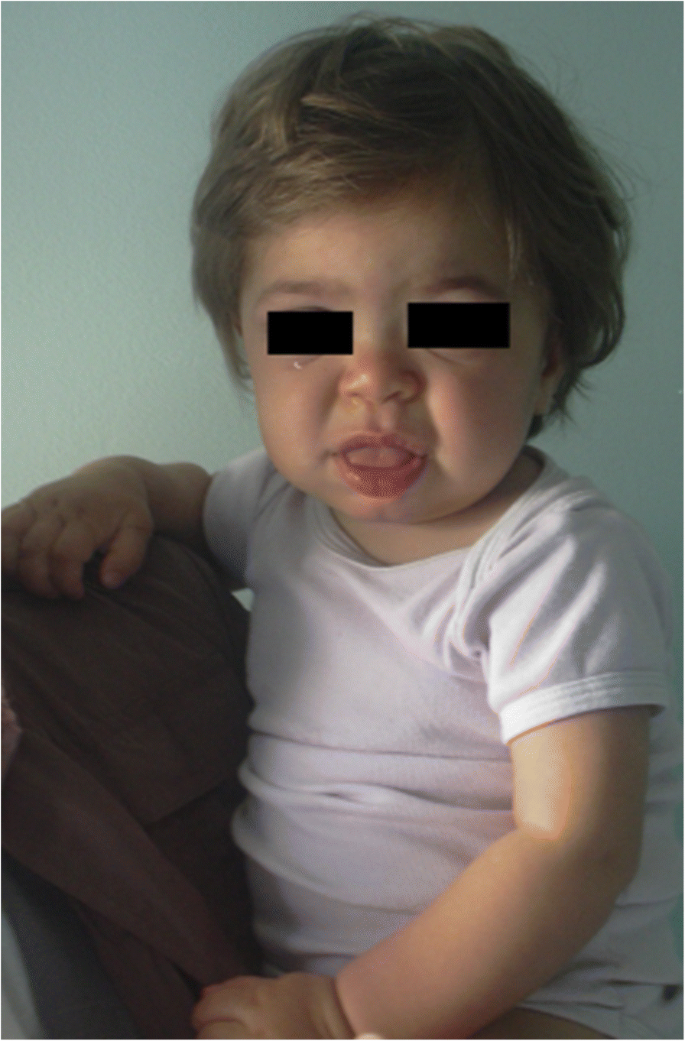
MPS IH en paciente de 1 año de edad. Facies gruesas: puente nasal plano, macroglosia, bosquejo frontal
En resumen, las manifestaciones esqueléticas son típicas y a veces precoces. Las facies gruesas y el retraso cognitivo no son signos prominentes tempranos.
¿Cuáles son las diferentes presentaciones de los diversos tipos de MPS?
MPS con afectación somática y cognitiva
Mucopolisacaridosis tipo I (MPS I; OMIM #252800) (Figs. 1, 2, 3, 4 y 5) es causada por una deficiencia de la hidrolasa lisosomal α-l-iduronidasa, que conduce a la acumulación multisistémica de dos GAGs: sulfato de dermatán (DS) y sulfato de heparán (HS) . En función de su gravedad, la MPS I se divide tradicionalmente en tres fenotipos clínicos; sin embargo, estas formas representan un continuo de gravedad de la enfermedad. El síndrome de Hurler, la forma más grave, se presenta típicamente durante el primer año de vida. Los niños afectados desarrollan rápidamente un deterioro cognitivo significativo y una enfermedad somática en múltiples sistemas de órganos, lo que lleva a la muerte en la primera década en ausencia de tratamiento. Las formas atenuadas de MPS I, conocidas como síndrome de Hurler–Scheie y síndrome de Scheie, respectivamente, se caracterizan por un inicio más tardío de los síntomas, una esperanza de vida más larga y una afectación leve o nula del sistema nervioso central (SNC). La herencia es autosómica recesiva, y en al menos el 50% de los casos es posible predecir el fenotipo sobre la base del genotipo, mientras que en el 50% restante se detectan nuevas mutaciones . La prevalencia es de aproximadamente 1 de cada 100.000 nacidos vivos .

MPS IH. un gibus en un paciente de 9 meses. radiografía de columna b de la columna vertebral en un paciente de 3 años de edad. c Imagen de resonancia magnética ponderada en T2 de la columna vertebral que muestra cifosis y estenosis del canal vertebral significativas

MPS IH. Pruebas radiográficas de disostosis múltiple. a Engrosamiento de las costillas en un paciente de 3 años y b displasia de cadera en otro paciente de 18 meses

MPS IH: a Mano con garra en un paciente de 18 meses de edad; b Radiografía de la mano.

MPS IH. Engrosamiento del hueso cortical del cráneo y conformación anormal en forma de “J” de la sella turca. Dilatación de los espacios periventriculares en imágenes de resonancia magnética ponderada b FLAIR y c T-2 de un paciente de 17 meses
Mucopolisacaridosis tipo II( MPS II; OMIM #309900) (Fig. 6), también conocido como síndrome de Hunter, es el resultado de la deficiencia de la enzima iduronato-2-sulfatasa (I2S) con la consiguiente acumulación de GAG de DS y HS . La prevalencia es de aproximadamente 1 de cada 140.000 a 156.000 varones nacidos vivos . En particular, esta es la única MPS ligada al cromosoma X, y las portadoras femeninas suelen ser asintomáticas; sin embargo, pueden verse excepcionalmente afectadas debido a anomalías del cromosoma X, homocigosis o inactivación sesgada del cromosoma X. La expresión fenotípica también abarca un amplio espectro de gravedad clínica. En los casos graves, coexisten el compromiso neurológico progresivo marcado y la enfermedad somática, y la muerte generalmente ocurre en la segunda década de la vida. Otros pacientes con disfunción neurológica mínima o nula pueden presentar un compromiso somático grave, mientras que, en el extremo opuesto del espectro, hay pacientes con manifestaciones somáticas mínimas e inteligencia normal que sobreviven hasta la edad adulta . Los signos y síntomas tempranos son compartidos por las formas graves de MPS I y II.

MPS II. Paciente de 3 años con rasgos faciales característicos leves
Los casos esporádicos de hidropesía fetal se describen en MPS I, aunque, en general, estos niños parecen normales al nacer. Los primeros signos de MPS I suelen aparecer durante los primeros meses de vida, mientras que los de MPS II suelen aparecer un poco más tarde . Kiely et al. , en un estudio retrospectivo en 55 pacientes con Hurler, observó que los síntomas respiratorios, las dificultades de alimentación, la hernia inguinal y la otitis media habían aparecido a una mediana de edad ≤ 6 meses, y cifosis, anomalías cardíacas, aumento del perímetro cefálico, hipotonía, organomegalia, opacidad corneal, restricción articular y hernia umbilical aparecieron entre 6 y 12 meses. La hernia umbilical es una de las primeras características de presentación en MPS I y II, y las hernias inguinales se notifican en aproximadamente el 60% de los pacientes .
Otra pista inicial común para MPS I y II puede ser infecciones recurrentes de las vías respiratorias superiores con aumento de secreciones. La otitis frecuente, la rinitis crónica recurrente y la secreción nasal persistente sin infección obvia no son específicas, pero pueden ayudar a fortalecer la sospecha diagnóstica si se asocian con signos más específicos. El almacenamiento de mordaza dentro de la orofaringe conduce al agrandamiento de las amígdalas y las adenoides, y contribuye a las complicaciones de las vías respiratorias superiores. Otro hallazgo frecuente es la respiración ruidosa, especialmente por la noche, asociada en las etapas posteriores con apnea obstructiva del sueño. Como consecuencia de las frecuentes infecciones del oído medio y la disostosis de los huesecillos del oído medio, la pérdida de audición es frecuente, con déficits conductivos y neurosensoriales . La traqueobroncomalacia se observa comúnmente y puede conducir a obstrucción aguda o colapso de las vías respiratorias, siendo esta una de las principales causas de muerte en los años posteriores .
La adenoidectomía, la amigdalectomía y la timpanostomía, junto con la reparación de hernia, son las intervenciones quirúrgicas más frecuentes y tempranas para los pacientes con MPS I y II, a menudo realizadas antes de conocer el diagnóstico (en más del 50% de los casos) .
La diarrea recurrente es bastante común en las primeras etapas de la MPS I, y también puede estar presente en pacientes con MPS II con afectación neurológica, mientras que es poco frecuente en los pacientes ligeramente afectados .
Deformidad gibosa (cifosis dorso-lumbar) (Fig. 2) se hace clínicamente evidente a una edad media de 8 años.6 meses a 1 año en MPS I, lo que representa un marcador bastante específico y precoz de los MPS en general. Los cuerpos vertebrales aparecen aplanados y con pico, lo que puede llevar a complicaciones posteriores, como atrapamiento del nervio espinal, lesión espinal aguda e inestabilidad atlanto-occipital; por lo tanto, se debe tener especial cuidado para evitar la dislocación de la articulación atlanto-axial durante la anestesia general y la cirugía. Después de 1 año de edad, las contracturas articulares y el genu valgum representan otros signos frecuentes de deterioro articular y displasia esquelética progresiva, que son fácilmente detectables antes de los 2 años de edad en todos los lactantes afectados por MPS. Todas estas manifestaciones esqueléticas juntas, típicas de la MPS, se denominan disostosis múltiple. El engrosamiento de las costillas se puede ver en las radiografías (Fig. 3a), los huesos largos son cortos con ejes anchos, y las rodillas son propensas a deformidades de varo y valgo. La disostosis falangeal y el engrosamiento sinovial conducen a una deformidad característica de la mano en la garra (Fig. 4), que es otra característica típica que puede levantar sospechas de la enfermedad. La displasia de cadera es con frecuencia la razón por la que estos niños pueden ser vistos por primera vez por especialistas ortopédicos. Por lo general, la pelvis está mal formada, las cabezas femorales son pequeñas y la coxa valga es común, con deformidad de cadera progresiva y debilitante (Fig. 3b).
Hay una diferencia notable entre MPS I y MPS II; en MPS I, el crecimiento esquelético se desacelera a los 3 años de edad , mientras que los pacientes con MPS II muestran un crecimiento excesivo en los primeros 5-6 años de edad, desacelerando su crecimiento a partir de entonces .
El engrosamiento de los rasgos faciales (nariz ancha con fosas nasales acampanadas, crestas supraorbitales prominentes, mejillas redondeadas grandes, labios gruesos, lengua protuberante agrandada) causado por el almacenamiento de mordazas en los tejidos blandos de la región orofacial y los huesos faciales se hace evidente en los primeros 2 años, a una edad media de 1,1 años para la enfermedad de Hurler y entre 18 meses y 4 años en la forma grave de la enfermedad de Hunter , aunque se pueden detectar cambios fenotípicos sutiles antes (ya en la segunda mitad primer año) por pediatras con experiencia específica (Fig. 1). La macrocefalia se hace progresivamente más evidente, y la escafocefalia es común (Fig. 5), pero la microcefalia también es posible en pacientes con Hurler . El hirsutismo facial y corporal siempre es evidente a la edad de 24 meses . La piel puede estar engrosada e inelástica. En particular, algunos pacientes con MPS II desarrollan una lesión cutánea distintiva, que se describe como pápulas de color blanco marfil (en forma de guijarros) en la parte superior de la espalda y los lados de la parte superior de los brazos, patognomónicas del síndrome de Hunter .
La miocardiopatía y el engrosamiento y rigidez progresivos de las valvas de las válvulas en pacientes con MPS I pueden llevar a regurgitación mitral y, con menor frecuencia, aórtica . Se puede detectar un soplo en los primeros meses de vida y, a veces, se ha notificado como el primer signo de la enfermedad . La afectación cardíaca también está presente en casi todos los pacientes con MPS II, pero esto comienza alrededor de los 5 años de edad . La valvulopatía puede llevar a hipertrofia ventricular derecha e izquierda e insuficiencia cardíaca.
La protuberancia del abdomen causada por hepatoesplenomegalia progresiva es fácilmente evidente después de los 6 meses de edad, aunque el almacenamiento de GAGs en el hígado y el bazo no conduce a disfunción orgánica .
La opacidad corneal se presenta en casi todos los individuos con MPS I antes de los 15 meses de edad , lo que puede causar una discapacidad visual grave. Por el contrario, la opacidad corneal no es una característica típica del síndrome de Hunter , pero el examen con lámpara de hendidura puede revelar lesiones corneales discretas que no afectan la visión. Se puede detectar disfunción retiniana, mientras que el glaucoma no es un hallazgo común.
El desarrollo psicomotor temprano generalmente se detecta como normal, pero en MPS I el retraso del desarrollo suele ser obvio a la edad de 18 meses, con un deterioro mental progresivo que conduce a una discapacidad intelectual grave. Las habilidades lingüísticas son muy limitadas en estos niños. Un retraso global de los hitos de desarrollo en MPS II se hace evidente a partir de 2 años . Sin embargo, la característica neurológica principal de la enfermedad de Hunter grave está representada por alteraciones del comportamiento marcadas, como hiperactividad, obstinación y agresividad, que normalmente no se observan en aquellos con el fenotipo atenuado .
La mucopolisacaridosis de tipo VII (MPS VII; OMIM # 253220), también conocida como síndrome de Sly, es una MPS ultra rara caracterizada por una actividad deficiente de la β-glucuronidasa, con almacenamiento lisosomal de sulfato de condroitina (CS), DS y HS, que conduce a disfunción celular y orgánica. El primer paciente fue descrito por Sly et al. en 1973 se han reportado en la literatura un total de 145 pacientes hasta la fecha.
La presentación clínica y la progresión de la enfermedad de MPS VII abarcan un amplio espectro de gravedad, desde manifestaciones tempranas, graves, multisistémicas y muerte en los primeros meses, hasta un fenotipo más leve con una inteligencia de aparición tardía, normal o casi normal, y una supervivencia más prolongada .
Las características fenotípicas de los pacientes con MPS VII se asemejan a las de MPS I y MPS II (estatura baja, displasia esquelética, macrocefalia, hipertrofia gingival, infecciones de oído recurrentes, hepatoesplenomegalia, hernias, afectación cardíaca, disminución de la función pulmonar y deterioro cognitivo). Una proporción inesperadamente alta de los pacientes descritos (41%) tiene antecedentes de hidropesía fetal neonatal no inmunitaria (NIHF) . A pesar de la aparición prenatal de la enfermedad en estos pacientes, 13 de cada 23 sobrevivieron a la infancia con un curso de leve a intermedio hasta finales de la adolescencia. Por lo tanto, la presencia de NIHF no predice, por sí sola, la posible gravedad del curso clínico si el paciente sobrevive a la infancia. Otro signo temprano es la macrocrania, mientras que la afectación de las válvulas cardíacas y las infecciones repetidas de las vías respiratorias superiores e inferiores aparecen en los primeros 2 años de vida. El retraso mental moderado y la pérdida auditiva progresiva con discapacidad del habla también son evidentes con el tiempo.
En resumen, las formas graves de MPS I tienen un inicio multisistémico muy temprano (dentro de los 6 meses de vida); La MPS II es similar, pero con un inicio posterior, y la MPS VII tiene un inicio prenatal con FNI frecuente y compromiso temprano grave del tracto respiratorio superior e inferior.
MPS con afectación principalmente neurológica y cognitiva
Mucopolisacaridosis tipo III (MPS III, también conocida como síndrome de Sanfilippo; Fig. 7) tiene una presentación neurológica prevalente. La MPS III es un trastorno de almacenamiento lisosomal causado por la deficiencia de una de las cuatro enzimas involucradas en el catabolismo del HS. Se conocen cuatro subtipos diferentes: MPS III tipo A (OMIM #252900), tipo B (OMIM #252920), tipo C (OMIM #252930) y tipo D (OMIM #252940), cada uno debido a una deficiencia enzimática diferente: tipo A, gen sulfamidasa/SGSH; tipo B, gen alfa—N—acetilglucosaminidasa/NAGLU; tipo C, alfa-glucosaminida N-acetiltransferasa/Gen HGSNAT; tipo D, gen N-acetilglucosamina-6-solfato sulfatasi/GNS). Los cuatro tipos (A a D) tienen herencia autosómica recesiva. La MPS III es la más frecuente de las MPS con una prevalencia estimada entre 0,3 y 4.1 casos por cada 100.000 recién nacidos, según el subtipo y la raza .

MPS IIIA. El mismo paciente a 2,5 y b 5 años de edad. Observe la expresión facial diferente, que muestra la progresión del deterioro neurológico
Los pacientes con síndrome de Sanfilippo presentan deterioro cognitivo progresivo en el segundo y tercer año de vida. En contraste con la mayoría de las MPS, las características somáticas son relativamente menos evidentes y el compromiso del SNC es visible. El retraso en el desarrollo del habla suele ser el primer signo notado por los padres, pero las principales preocupaciones pueden ser problemas de comportamiento (hiperactividad, comportamiento ansioso y agresivo, trastornos del sueño), seguidos de un deterioro mental progresivo . Estos síntomas pueden causar diagnósticos erróneos como trastornos del comportamiento, trastorno por déficit de atención con hiperactividad (TDAH) y trastornos del espectro autista . La epilepsia también puede estar presente.
Los trastornos del sueño, cuya incidencia se reporta hasta un 80-90%, son una característica común. Consisten en dificultades para conciliar el sueño y despertar nocturno frecuente, con una inversión completa del ritmo día–noche en algunos pacientes.
Todas estas manifestaciones son muy difíciles de manejar y tienen un enorme impacto psicológico en la calidad de vida de toda la familia.
En general, aunque el fenotipo puede ser muy similar en los cuatro subtipos, el curso clínico en Sanfilippo B y C parece caracterizarse por un fenotipo menos grave .
Los síntomas no neurológicos suelen ser menos pronunciados en la MPS III que en las otras MPS. Las infecciones recurrentes de oído, nariz y garganta se observan a una edad más temprana. La otitis recurrente, los defectos óseos del oído medio y las anomalías en el oído interno pueden causar sordera. Un exceso de secreciones gruesas y cambios anatómicos pueden producir obstrucción de las vías respiratorias. En los primeros años de vida, se notifican con frecuencia diarrea, mientras que el estreñimiento es más frecuente en pacientes de edad avanzada.
El examen físico puede mostrar rasgos faciales gruesos, aunque estos son menos pronunciados. Los pacientes tienen macrocefalia, cejas anchas con llamaradas mediales y sinofrias, el cabello generalmente es seco y grueso, y la hipertricosis es común. En pacientes más jóvenes, se observan con frecuencia hepatomegalia leve y hernias umbilicales e inguinales. Las contracturas raras, y a menudo leves, se encuentran principalmente en los codos. El crecimiento se ve afectado en aproximadamente la mitad de los pacientes mayores de 12 años .
En resumen, la enfermedad de MPS III o Sanfilippo tiene una afectación neurológica prominente con signos somáticos muy leves. Se puede sospechar que los niños de 2 a 3 años de edad que desarrollan hiperactividad, TDAH y comportamiento agresivo tienen MPS III.
MPS con afectación somática prevalente
MPS IV y MPS VI se presentan solo con afectación somática. Estas son condiciones progresivas que ahorran capacidad intelectual y afectan principalmente al esqueleto (MPS IVA), o a todos los órganos/sistemas del cuerpo (MPS VI). La herencia es autosómica recesiva. La velocidad a la que los síntomas empeoran varía entre los individuos afectados.
Mucopolisacaridosis IV (MPS IV, también conocido como síndrome de Morquio; Fig. 8) se presenta como dos trastornos genéticamente distintos, cada uno con una deficiencia enzimática diferente: MPS IVA (síndrome de Morquio A; OMIM #253000) debido a la deficiencia de N-acetilgalactosamina-6-sulfatasa (gen GALNS), lo que resulta en la acumulación de queratán-sulfato (KS) y CS ; y MPS IVB (síndrome de Morquio B; OMIM #253010) debido a la deficiencia de la actividad de la beta-galactosidasa que conduce a la excreción de oligosacáridos y KS en orina como en GM1 pacientes. MPS IVB es de hecho alélico a las diversas formas de gangliosidosis GM1, pero carece del deterioro psicomotor visto en la gangliosidosis GM1 .

MPS IVA. paciente de 4 años con radiografías b anteroposterior y c lateral que muestran costillas en forma de paleta y vértebras aplanadas y redondeadas con un aspecto típico de “pico anterior”
Los pacientes con MPS IVA grave sin actividad enzimática detectable revelan sus primeros síntomas generalmente durante el primer año de vida, aunque rara vez se diagnostican antes de los 4-5 años de edad. La información sobre la historia natural de los pacientes de Morquio A se deriva principalmente de dos colecciones de datos de gran alcance . La tasa de crecimiento reducida comienza aproximadamente a los 18 meses de edad y el crecimiento se detendrá aproximadamente a los 7 u 8 años de edad. A diferencia de las otras MPS, las articulaciones de Morquio A suelen ser laxas y muy flexibles (hipermóviles), pero algunas articulaciones (generalmente las caderas y los hombros) pueden tener un rango de movimiento restringido. Los pacientes con tal compromiso esquelético pueden recibir un diagnóstico o someterse a una evaluación de displasia espondiloepifisaria, pseudoacondroplasia, displasia epifisaria múltiple o enfermedad bilateral de Legg-Calvé-Perthes .
Una característica constante es la hipoplasia odontoide; por lo tanto, la dislocación de la articulación atlanto-occipital puede ocurrir causando compresión y daño a la médula bulbar y espinal, lo que resulta en parálisis o incluso la muerte si la estabilización cervical no se realiza a tiempo.
Los pacientes con MPS IVB tienen una manifestación clínica muy similar, pero con una estatura baja menos prominente. Tienen rasgos faciales ligeramente toscos, pérdida de audición, opacidades corneales, cardiopatía valvular e infecciones frecuentes del tracto respiratorio superior. También pueden presentarse con hernia inguinal y hepatomegalia leve. Las características displásicas esqueléticas incluyen platispondilia, hipoplasia odontoide con posible subluxación cervical, cifoescoliosis, coxa valga y alas ilíacas constrictas .
Mucopolisacaridosis VI (MPS VI, también conocida como síndrome de Maroteaux-Lamy; OMIM # 253200; Fig. 9) se debe a mutaciones patógenas en el gen de la arilsulfatasa B (ARSB) ubicado en el cromosoma 5. Como consecuencia, la actividad reducida o ausente de la enzima arilsulfatasa B (ASB) (N-acetilgalactosamina 4-sulfatasa) deteriora la degradación del DS determinando su acumulación celular .

MPS VI. Un niño de 2 años con evidente cara gruesa y disostosis esquelética
Los pacientes sin actividad enzimática detectable tienen la forma grave de MPS VI y presentan síntomas en la primera infancia. Su fenotipo somático es similar al síndrome de Hurler. En la mayoría de los casos, a los 2 o 3 años de edad, el daño esquelético severo y progresivo, llamado disostosis múltiple, se hace evidente. Esta displasia esquelética incluye displasia de cadera con cabeza femoral displásica, cuerpos vertebrales anormales, clavículas irregulares, cúbito y radio distales hipoplásicos, y huesos metacarpianos displásicos y cortos; los huesos carpiano y tarso son hipoplásicos y tienen un perfil irregular. La velocidad de crecimiento a menudo se ralentiza después del primer año de vida, con una altura final generalmente inferior a 120 cm. Al igual que en otros MPS, un signo temprano son los rasgos faciales gruesos. Además, los pacientes tienen un abdomen sobresaliente típico, hepatomegalia, hernia umbilical y/o inguinal y compromiso cardíaco en la primera infancia. Aunque no se presenta discapacidad intelectual primaria, se puede observar hidrocefalia en algunos pacientes con la consiguiente hipertensión intracraneal y papiledema. El Pectus carinatum, las articulaciones rígidas y contraídas, la escoliosis y cifosis, y la compresión de la médula espinal cervical empeoran con el tiempo .
En resumen, las MPS IV y VI no muestran compromiso del SNC. MPS IV es un único MPS en mostrar laxitud articular, mientras que el inicio severo de MPS VI es similar al observado en MPS I.