Autosomaalinen resessiivinen synnynnäinen iktyoosi / Actas Dermo-Sifiliográficas
Johdanto
viimeisin konsensusluokitus iktyoosi erottaa toisistaan kaksi päämuotoa: ei-syndromiset muodot, joissa esiintyy vain iho-oireita, ja syndromiset muodot, jotka esiintyvät ilmentyminä myös muissa elimissä (Taulukko 1).1 joukossa nonyndromic muotoja, 4 ryhmät tunnistetaan: yhteinen ichthyoses, autosomaalinen resessiivinen synnynnäinen ichthyoses (ARCIs), keratinopathic ichthyoses, ja muita harvinaisempia ichthyoses.Perinteisesti ryhmä ARCIs oli jaettu 2 häiriöt, lamellar ichthyosis (LI) ja synnynnäinen ichthyosiform erytroderma (CIE). Uudessa luokituksessa Harlequin ichthyosis (HI) lisättiin tähän ryhmään 1,koska inaktivoivat mutaatiot ABCA12-geenissä on todettu aiheuttavan tämän häiriön,2,3 kun taas nonsense-mutaatiot samassa geenissä voivat aiheuttaa LI4-tai CIE5, 6-fenotyypin. Muita harvinaisempia ARCIs-ryhmään kuuluvia variantteja ovat itsensä parantava collodion baby (SHCB), acral SHCB ja bathing suit ichthyosis.7-9
konsensus luokittelu perustuu kliinisiin ominaisuuksiin Ichthyosis1.
| Ei -dromiset muodot | Syndromiset muodot |
| Common IchthyosesIchthyosis vulgarisRecessive x-linkitetty ichthyosis (nonyndromic )ajimajor muotosharlekiini ichthyosisLamellar ichthyosisCongenital ichthyosiform erythrodermaMinor formsSelf-healing kollodium babyAcral self-healing kollodium babyBathing suit ichthyosisKeratinopathic IchthyosesMajor formsepidermolyyttinen Ichthyosissupicial epidermolyyttinen ichthyosisminor formsannulaarinen epidermolyyttinen ichthyosiscurth-Macklin Ichthyosisautosomaalinen resessiivi epidermolyyttistä ichthyosisepidermolyyttistä luovaa muuta muotoa Klorikriini keratodermaErythrokeratodermia vararabilispeeling skin syndromekongenitaalinen retikulaarinen ichthyosiform erytrodermaklick syndrome | Syndrominen X-linkitetty Ichthyosrecessiivinen x-linkitetty ichthyosis (syndrooma)Ichthyosis follicularis, alopecia, and photophobia (IFAP) syndromekonradi-Scammann-happle syndrome (chondrodysplasia punctata)tyyppi 2) syndrooma autosomaalinen ichthyosisskin disordersneterton syndromeichthyosis-hypothrichosis syndromeichthyosis-sklerosoiva kolangitis syndrometrichothystrofyneurologinen disorderssjögren-Larsson syndromeRefsum diseaseMEDNIK syndromeFatal disease courseGaucher disease, type 2Multiple sulfatase deficiencyCEDNIK syndromeARC syndromeOther associated signsKID syndromeChanarin-Dorfman syndromeIchthyosis prematurity syndrome |
Abbreviations: ARC, arthrogryposis–renal dysfunction–cholestasis; ARCI, autosomal recessive congenital ichthyosis; CEDNIK, cerebral dysgenesis, neuropathy, ichthyosis, and palmoplantar keratoderma; KID, keratitis ichthyosis deafness; KLICK, keratosis linearis with ichthyosis congenital and sclerosing keratoderma; MEDNIK, kehitysvammaisuus, enteropatia, kuurous, perifeerinen neuropatia, iktyoosi, keratoderma.
Arcisin epidemiologiasta on vain vähän tietoa. Yhdysvalloissa LI: n levinneisyydeksi on arvioitu syntyessään 1 100 000 asukasta kohti ja CIE: n 1 200 000 asukasta kohti. Muissa tutkimuksissa LI: n ja CIE: n yhteenlasketuksi esiintyvyydeksi on ilmoitettu 1 200 000-300 000 asukasta kohti.10,11 joissakin maissa, kuten Norjassa, arvioitu esiintyvyys on suurempi (1 / 91 000) perustajamutaatioiden vuoksi.12 yhden tai useamman toistuvan mutaation löytäminen populaatiossa voi johtua siitä, että mutaatio tapahtui tietyssä historian vaiheessa ja siirtyi sukupolvelta toiselle (perustajamutaatio) tai siitä, että genomin alueella, josta mutaatio löytyy, on mutaatiolle altis DNA-sekvenssi (mutaatiopesäke). Espanjassa ARCI: n arvioitu esiintyvyys on 1 / 138 000 koko väestöstä ja 1 / 61 700 alle 10-vuotiaista lapsista.13 tietyillä Espanjan alueilla esiintyvyys saattaa olla vielä suurempi. Esimerkiksi Galician rannikolla esiintyvyyden ilmoitettiin olevan 1 per 33 000, mikä johtui myös perustajavaikutuksesta.14
Lamellaarinen iktyoosi ja synnynnäinen Iktyosiforminen Erytrodermakliiniset ominaisuudet
vaikka alun perin luultiin, että LI ja CIE ovat eri kokonaisuuksia, on raportoitu potilaista, joilla on väli-kliinisiä ilmenemismuotoja, ja molemmat tilat voivat johtua mutaatioista samassa geenissä.15,16 lisäksi potilaat, joilla on sama mutaatio, jopa saman perheen sisällä, voivat kehittää erilaisia fenotyyppejä.12, 15
useimmat potilaat syntyvät kollodiumkalvon ympäröiminä, joka vähitellen häviää ensimmäisten elinviikkojen aikana ja korvautuu lopullisella fenotyypillä (Fig. 1 A). Hypohidroosi, vaikea lämpö intoleranssi, ja kynsien dystrofia ovat usein havaittu sekä LI ja CIE.17-19 LI-potilaalla on yleensä vaikeampia kliinisiä oireita kuin CIE-potilailla. Niillä on suuret platelimaiset, usein tumman väriset suomut, jotka peittävät koko ruumiin pinta-alan. Erytrodermia on joko puuttuu tai minimaalinen. Tällaisilla potilailla on yleensä ektropion ja, joskus, eclabium, hypoplasia nivel-ja nenän ruston, arpia hiustenlähtö, erityisesti reunalla päänahan, ja palmoplantar keratoderma (Fig. 1B ja C). CIE: lle on ominaista erytrodermian esiintyminen ja hieno valkeahko skaalaus (Kuva. 2). Joillakin potilailla on havaittavissa eryteemaa ja yleistynyttä hilseilyä. Suomut voivat olla suuria ja tumman värisiä erityisesti jalkojen ojentajapinnoilla. Lievemmissä tapauksissa eryteema on lievä ja hilseily on kunnossa.

Lamellaarisen iktyoosin kliiniset piirteet. A, ruskehtava lamellimainen desquamaatio. B, merkitty plantaarihyperkeratoosi. C, arpeutuminen hiustenlähtö päänahan.
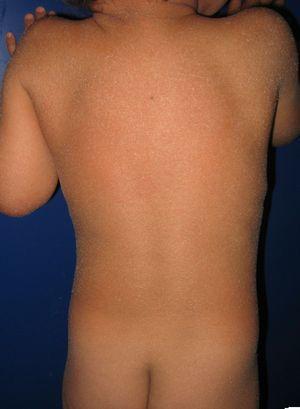
potilas, jolla on synnynnäinen ichthyosiform erytroderma ja mutaatioita ALOXE3-geenissä. Lievä eryteema ja yleistynyt vaalea furfuraceous desquamation voidaan nähdä.
histopatologia
histopatologisia muutoksia ei voida diagnosoida. LI: ssä havaitaan massiivista ortokeratoottista hyperkeratoosia, jossa on yleensä kaksi kertaa enemmän laajentumaa kuin CIE: ssä. Orvaskesi on akantoottinen ja saa toisinaan psoriasiksen kaltaisen vaikutelman. Solujen proliferaatio on normaali tai hieman kohonnut.CIE: tä sairastavilla 17-19 potilaalla on vähemmän huomattava hyperkeratoosi, fokaalinen tai laaja parakeratoosi, normaali tai paksuuntunut rakeinen kerros ja voimakkaampi akantoosi. Epidermaalinen vaihtuvuus kasvaa.17-19
Ultrarakenne
vaikka tähän mennessä ei ole löydetty läheistä korrelaatiota molekulaaristen, kliinisten ja ultrarakenteisten löydösten välillä, elektronimikroskopia voi kuitenkin olla hyödyllinen muiden iktyoosin muotojen poissulkemiseksi ja geneettisten analyysien ohjaamiseksi joissakin tapauksissa. Synnynnäistä iktyoosia on kuvattu neljää tyyppiä (Taulukko 2).
Ultrastructural luokittelu synnynnäinen Ichthyoses.
| Tyyppi | pääpiirre | muut ominaisuudet | mutaatiot | kliiniset oireet |
| 1 | iktyoosityyppien 2, 3 ja 4 ultrarakenteisten merkkiaineiden poissaolo | Lipidipisarat tai-renkaat marraskeden (useimmin)pienissä keratohyaliinisissa granulesvesikulaarisissa tai lobulaarisissa kalvopäällysteisissä rakeissa | TGM1 (33.3%) ALOX12B (2 tapausta) | CIE |
| 2 | Kolesterolihuokoset marraskedessa | reunustettujen vaipojenpienet keratohyaliinin granuleslipidipisarat | TGM1 (89-100%) | LI |
| 3 | Laminoidut kalvorakenteet granulosum-ja/tai marraskeden kerrostumissa. | epänormaali kalvopäällyste granuleslipidipisaratfoci merkittäviä rinnakkaisia tumakuoleja rakeisessa kerroksessa | NIPAL4 (93%) | CIE (yleisin)LI |
| 4 | Trilamellaarikalvopakkaukset, jotka täyttävät joitakin soluja granulosum-ja/tai marraskeden kerrostumassa | epänormaalit kalvopäällystysrakeet | FTAP4 | iktyoosi keskosoireyhtymä (100%) |
lyhenteet: CIE, synnynnäinen ichthyosiform erythroderma; LI, lamellar ichthyosis.
synnynnäinen iktyoosi tyyppi 1
synnynnäinen iktyoosi tyyppi 1 on ominaista ultrarakenteisten markkereiden puuttuminen iktyoosi tyypit 2, 3, ja 4. Siksi diagnoosi tehdään yleensä vasta, kun muut tyypit on suljettu pois. Yleisin havainto on rasvapisaroiden tai renkaiden esiintyminen marraskeden (Kuva. 3 A).20 nämä lipidipisarat eivät ole vakio ominaisuus tai spesifisiä tälle nimenomaiselle tyypille, koska niitä ei ole kaikissa tapauksissa,20 ja niitä voi esiintyä muunlaisissa iktyooseissa.21, 22 kliinisesti useimmilla potilailla esiintyy CIE: n ilmentymiä.12,20 kolmasosalla potilaista on mutaatioita TGM1-geenissä.16 tämä ultrarakenteinen tyyppi on myös tunnistettu alox12b-geenin mutaatioiden yhteydessä.23,24

Elektronimikroskooppikuvat. A, synnynnäinen iktyoosi tyyppi 1, osoittaa lipidipisarat marraskeden ja puuttuminen ultrarakenne markkereita muiden tyyppien iktyoosi. B, synnynnäinen ichthyosis tyyppi 2, ominaista läsnäolo kolesteroli halkeamat (nuoli) sarveiskalvon.
synnynnäinen iktyoosi tyyppi 2
synnynnäinen iktyoosi tyyppi 2 on ominaista kolesteroli halkeamat marraskeden (Kuva. 3b).21 tällaiset halkeamat ovat jatkuva havainto tämän tyyppisessä iktyoosissa, ja ne voidaan havaita eri koepaloissa samassa potilaassa; suun retinoidien hoidolla ei ole vaikutusta näihin halkeamiin.12,25 Elektronitiheää aggregaattia on havaittu myös sarveissyyteissä joillakin potilailla, joilla on puutteellinen TGase 1-aktiivisuus.26-28 kliinisesti useimmilla potilailla on vakavia CIE: n ilmenemismuotoja.12 tämä ultrarakenteinen tyyppi liittyy vahvasti TGM1-geenin mutaatioihin.12,16
synnynnäinen iktyoosi tyyppi 3
synnynnäinen iktyoosi tyyppi 3 on ominaista lamellaarinen kalvorakenne granulosum-ja/tai marraskeden kerrostumassa. Nämä rakenteet ovat järjestäytyneet liuskoiksi tyhjän tilan ympärille lähelle ydintä.22,29-31 tämän tyypin kliiniset ilmenemismuodot ovat erilaisia kuin muut; iktyoosin puhkeaminen vaihtelee, hilseily ja punoitus voivat olla hajanaisia tai yleistyneitä, ja erityisesti taivutukset vaikuttavat. Nipal4-geenin mutaatiot aiheuttavat 93% tyypin 3 iktyooseista.32
synnynnäinen iktyoosi Tyyppi 4
tyypillisesti synnynnäisessä iktyoosissa tyyppi 4 jotkut granulosum-ja marraskeden solut ovat täynnä trilamellaarisia kalvopakkauksia.33 nämä löydökset ovat patognomisia iktyoosin keskosoireyhtymälle, jota pidetään tällä hetkellä iktyoosin syndroomisena muotona.34,35
Molekyylitutkimukset
geneettiseltä kannalta Arcit ovat hyvin heterogeenisiä. TGM1-geeni liittyy useimpiin tapauksiin, mutta mutaatioita on raportoitu 5 muussa geenissä (ALOX12B, ALOXE3, NIPAL4, CYP4F22 ja ABCA12). Fischer ym.36 tutki 520 perhettä, joilla oli ARCI, ja havaitsi mutaatioita vähintään yhdessä näistä geeneistä 78%: ssa tapauksista (TGM1 32%: ssa, NIPAL4 16%: ssa, ALOX12B 12%: ssa, cyp4f22 8%: ssa, ALOXE3 5%: ssa ja ABCA12 5%: ssa). Toisessa tutkimuksessa, johon osallistui 250 eri alkuperää olevaa ARCI-potilasta, 38%: lla oli TGM1-mutaatioita, 6, 8%: lla ALOXE3-mutaatioita ja 6, 8%: lla ALOX12B-mutaatioita.37 Galiciassa tunnistimme mutaatioita TGM1 -, ALOX12B -, ALOXE3 -, NIPAL4-ja cyp4f22-geeneissä 75 prosentissa tutkituista perheistä, mutta mutaatioiden jakauma oli erilainen.14 TGM1-geeni mutatoitui vuonna 68.7% tapauksista, kun ALOXE3-geeni mutatoitui vain yhdellä potilaalla. Emme havainneet mutaatioita missään muussa tutkitussa 3 geenissä.
TGM1
TGM1-geeni sijaitsee kromosomissa 14q11.2 ja sillä on 15 eksonia (GenBank NM-000359.2). Se koodaa Tgaasi 1-entsyymiä, joka on yksi orvaskeden 3: sta Tgaasientsyymistä.38 tämä entsyymi osallistuu kornisoidun kuoren muodostumiseen katalysoimalla useiden proteiinien, kuten involukriinin, lorikriinin ja proliinipitoisten proteiinien, kalsiumriippuvaista ristisidontaa.39,40 se myös katalysoi sitomista ??- hydroksikeramideja reunustetun kuoren uloimmassa kerroksessa ja proteiineja sisemmässä kerroksessa.41, 42 potilailla, joilla on TGM1-mutaatio, cornified kirjekuori puuttuu ja TGase 1: n aktiivisuus on vähentynyt tai sitä ei ole lainkaan.43-47
vuodesta 1995 lähtien,jolloin tämän geenin todettiin aiheuttavan joitakin ARCI-tapauksia, eri alkuperää olevilla potilailla on raportoitu 48-50 yli 110 mutaatiota. Mutaatiot TGM1 ovat yleisin syy ARCI.36,37 tämä mutaatio on todettu 55%: ssa tapauksista Yhdysvalloissa ja 84%: ssa tapauksista Norjassa.12,51 yleisin mutaatio on c.877-2A>g, jota on löydetty 34%: sta tähän mennessä raportoiduista mutatoituneista alleeleista.52 tämän mutaation suuri yleisyys esimerkiksi Yhdysvalloissa ja Norjassa johtuu perustajavaikutuksesta.12,53 toiseksi yleisin mutaatio on P. Arg142His. Tätä ja vastaavia mutaatioita on raportoitu muun muassa Egyptissä, Saksassa, Suomessa ja Yhdysvalloissa 15, 49-51,54-56 ja näyttäisi siltä,että nämä ovat hotspot-mutaatioita.57 p.Arg307Trp-mutaatio on yleinen japanilaisessa väestössä.5 Galiciassa p. Arg760X, c.1223_1227delacaca ja c.984 + 1G>TGM1: n mutaatioita todettiin 81, 82%: ssa perheistä, joilla oli tämän geenin mutaatioita, mikä viittaa perustajavaikutukseen.14 vahvistus tämä hypoteesi saatiin haplotype tutkimus (työ vielä julkaisematon).
TGM1-mutaatiot ovat vastuussa useimmista li15-tapauksista,27,44,46,56,58-63 ja pieni osa tapauksista CIE.43,47,64,65 tällaiset mutaatiot voivat aiheuttaa myös muita kaaren muotoja, kuten SHCB: tä, acral SHCB: tä ja uimapuvun iktyoosia.
monissa tutkimuksissa on yritetty osoittaa genotyyppi-fenotyyppi-yhteyksiä TGM1: n mutaatioiden ja ultrarakenteisten tai kliinisten löydösten välillä, mutta merkittävää korrelaatiota ei ole toistaiseksi havaittu.15,16,53 potilaat, joilla on TGM1-geenin mutaatioita, kärsivät yleensä vakavammin kuin potilaat, joilla ei ole tällaisia mutaatioita. Tutkimuksessa, jossa oli mukana 83 ARCI-potilasta Ruotsissa ja Virossa, ektropionin ja kollodium-vauvan esiintyminen liittyi TGM1-mutaatioihin, kun taas eryteemaa havaittiin enemmän potilailla, joilla ei ollut mutaatioita tässä geenissä.66 toinen tutkimus osoitti, että skaalaustyyppi on tärkein ero TGM1-mutaatioiden kantajien ja ei-kantajien välillä, kun havaittiin, että kaikilla potilailla, joilla oli tämän geenin mutaatioita, oli lamellaarinen skaalaus, kun taas 80 prosentilla potilaista, joilla ei ollut TGM1-mutaatioita, oli hieno skaalaus.14 lisäksi on havaittu, että typistävät mutaatiot liittyvät useammin hypohidroosiin ja hikoiluhäiriöihin kuin missense-mutaatiot.51 Pohjois-Amerikan populaatiossa malli, joka perustuu tiettyjen kliinisten ominaisuuksien esiintymiseen, ennustaa, että potilailla, jotka ovat syntyneet kollodium-vauvoina ja joilla on silmäsairauksia ja/tai alopekiaa, on 4 kertaa todennäköisemmin TGM1-mutaatioita.51
ALOXE3 ja ALOX12B
aloxe3-ja ALOX12B-geenit sijaitsevat kromosomissa 17p13.1.67 niillä on samanlainen rakenne, jossa on 15 eksonia, jotka koodaavat epidermaalisia Loxeja eLOX-3 ja 12R-LOX.68,69 se, että ne ilmenevät pääasiassa orvaskeden suprabaalikerroksissa, tukee niiden roolia epidermaalisen erilaistumisen edistyneissä vaiheissa ja osallistuu lamellikappaleiden käsittelyyn.24,70 nämä entsyymit vaikuttavat hepoksiliinireitin viereisiin vaiheisiin (Kuva. 4). 12R-LOX muuttaa arakidonihapon 12R-hydroksieikosatetraeenihapoksi,kun taas eLOX-3 muuntaa tämän tuotteen hepoksiliini A3-perheen epoksialkoholi-isomeriksi 69, 71.72 hepoksiliinituote on epästabiili ja hydrolysoituu soluissa spesifiseksi trihydroksijohdannaiseksi (trioksiliini). Vaikka hepoksiliinireitin tuotteiden tarkkaa roolia ei tunneta, on arveltu, että ne voivat osallistua marraskeden solujenvälisten lipidien muodostumiseen tai toimia signaaleina, jotka indusoivat keratinosyyttien erilaistumista.
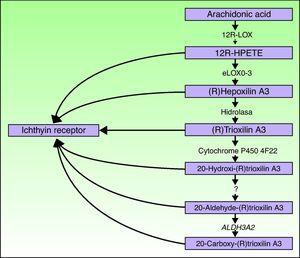
Hepoksiliinireitin kaavio, joka osoittaa aloxe3 -, ALOX12B -, NIPAL4-ja cyp4f22-geenien osallistumisen. Näiden geenien mutaatiot ovat vastuussa tietyntyyppisistä kaarista. HPETE viittaa hydroperoksieikosatetraeenihappoon.
ALOX12B-ja ALOXE3-geenit tunnistettiin ensimmäisen kerran 2002.73,74 sen jälkeen on raportoitu yli 30 mutaatiota ALOX12B-gene23, 24,37,75-77 ja noin 10 mutaatiota ALOXE3-gene37,74,75. Nämä mutaatiot ovat vastuussa 14-17% ARCIs36,37 ja 72.2% SHCBs: stä.23,78,79 näiden mutaatioiden ja fenotyypin välinen syy-yhteys vahvistettiin osoittamalla,että epidermaalisen LOXIN katalyyttinen aktiivisuus oli täysin hävinnyt potilailla, joilla oli nämä mutaatiot75, 80, ja käyttämällä eläinmalleja, jotka toistivat ihmisillä nähdyn iktyosiformisen fenotyypin.81-83 molemmat geenit ovat vastuussa samasta prosentista ARCI-tapauksia. ALOXE3-geenin eri mutaatioiden kirjo on kuitenkin rajallinen johtuen 2 mutaation, P.Arg234X: n ja P.Pro630Leu: n hallitsevuudesta, jotka näyttävät vastaavan hotspotteja.37, 74, 75
potilailla, joilla on mutaatioita ALOXE3-ja ALOX12B-geeneissä, on yleensä CIE-fenotyyppi.74,75,77 asteikon vaikeusaste on lievä tai kohtalainen, ja suomujen väri on vaalea tai vaaleanruskea. Eryteemaa voi myös esiintyä. Jopa 76% potilaista on syntynyt kollodium-vauvoina ja 88%: lla on hikoiluhäiriöitä.37 potilaalla, joilla oli alox12b-geenin mutaatioita, esiintyi vähäisempää, valkeahkoa desquamaatiota verrattuna ALOXE3-geenin mutaatioiden kantajiin. Tällöin suomut ovat ruskehtavia ja kiinnittyneitä. Eryteeman esiintyminen, palmoplantarin hyperkeratoosi ja palmoplantarin poimujen korostuminen liittyvät myös ALOX12B-mutaatioihin.37
Ichthyin / NIPAL4
nipal4-geeni, joka tunnetaan myös nimellä ichthyin-geeni, sijaitsee kromosomissa 5q33. Siinä on 6 eksonia, jotka koodaavat proteiinia, jolla on useita transmembraanisia domeeneja, joiden toiminta on tuntematon.84 on arveltu, että proteiinituote osallistuu samaan metaboliareittiin kuin LOX ja voi toimia reseptorina trioksiliineille A3 ja B3 tai muille hepoksiliinin metaboliareitin metaboliiteille.84 näin se olisi osallisena lamellikappaleiden muodostumisessa tai niiden kuljetuksessa kohti solunulkoista avaruutta.32 tätä tukevat 2 huomautusta. Ensinnäkin 93%: ssa tapauksista, mutaatiot tämän geenin liittyy ultrastructural malli synnynnäinen ichthyosis tyyppi 3, ominaista poikkeavuuksia lamellaaristen elinten ja läsnäolo pitkänomainen perinukleaarinen kalvot stratum granulosum.32 toiseksi, NIPAL4 ilmaistaan olennaisesti orvaskeden granulosum-kerrostumassa, jossa lamellaariset kappaleet ovat läsnä.85
sen jälkeen, kun NIPAL4-geeni löydettiin vuonna 2004, on raportoitu vain 9 mutaatiota Välimeren maista (Algeria,Turkki Ja Syyria), 84 Skandinavian maasta,32 Pakistanista,85 Färsaarilta, 32 ja Etelä-Amerikasta.84
niiden potilaiden kliininen kirjo, joilla on tämän geenin mutaatioita, on laaja, jopa saman perheen jäsenten keskuudessa. 3,7% 32-60% 84 syntyy kollodiumvauvoina. Kun kollodium-kalvo katoaa, useimmat potilaat kehittävät CIE: n ilmentymiä, joissa on hienoja vaaleita asteikkoja punoittavalla pohjalla kasvoissa ja rungossa ja suuremmat, ruskehtavat asteikot kaulassa, pakaroissa ja jaloissa.84 merkittyä kseroosia, yleistynyttä ruskehtavaa retikulaarista hyperkeratoottista plakkia, jotka näkyvät korostuneina ihopoimuissa, ja kasvojen dyskromia voi olla läsnä.32,85 lisäksi palmoplantar keratoderma on usein löydös yhdessä satunnaisten sormien kontraktuurien ja käyrien kynsien kanssa. Joissakin tutkimuksissa on raportoitu Lille tyypillisempiä löydöksiä.Atooppisen ihottuman merkkejä ja oireita on raportoitu joillakin potilailla, joskaan FLG-geenin mutaatioita ei havaittu yhdessäkään näistä tapauksista.85
cyp4f22
flj39501 – tai cyp4f22-geeni sijaitsee kromosomissa 19p13.12.86 sillä on 12 eksonsia87 ja se koodaa P450-sytokromia, sukua 4, alaheimoa F, polypeptidiä 2, leukotrieeni B4-ω-hydroksylaasin (CYP4F2) homologia. Ihossa olevan flj39501: n tuotteen ja reaktion substraattien katalysoima reaktio voidaan päätellä analogisesti sen tunnettujen homologien CYP4F2 ja cyp4f3 kanssa.88 on oletettu, että cyp4f2 ja cyp4f3 osallistuvat hepoksiliinireittiin katalysoimalla trioksiliini A3: n muuntumista 20-hydroksi-(R)trioksiliini A387: ksi ja että tämän reitin lopputuotteella, 20-karboksi-trioksiliini A3: lla, voi olla keskeinen biologinen säätelyvaikutus ihossa.89
tähän mennessä tämän geenin mutaatioita on raportoitu vain 8 kappaletta 12: ssa Välimeren maista peräisin olevassa verisukulaisperheessä87 ja 1: ssä israelilaista alkuperää olevassa perheessä.62
lefèvren ym. ilmoittamissa perheissä., 87 useimmilla potilailla oli syntyessään CIE: n fenotyyppi, ja tämä eteni myöhemmin LI: iin. potilaat olivat yleensä syntyneet merkityillä erytrodermioilla, joskin ilman kollodiumikalvoa. Vanhetessaan heille kehittyi yleistynyt valkeaharmaa skaalaus, joka oli selvempi periumbilisellä alueella, pakaroissa ja kehon alaosassa. Kämmenien ja jalkapohjien hyperlineaarisuus ja hiuspohjan hilseily olivat yleisiä pityriasiformisen tyypin aikoina.87 toisessa perheessä sairastuneet 3 jäsentä syntyivät collidion-vauvoina ja kehittyivät voimakkaaseen erytrodermiaan, yleistyneeseen hilseilyyn ja palmoplantaariseen keratodermaan.62
ABCA12
vuonna 2003 ABCA12-geenin raportoitiin aiheuttavan joitakin LI-tapauksia ja se kartoitettiin kromosomiin 2q34.4 myöhemmin vahvistettiin, että tämän geenin mutaatiot olivat vastuussa myös HI: stä.2, 3ABCA12 koodaa 53 eksonia ja kuuluu ABC-transportteriperheeseen, joka sitoo adenosiinitrifosfaattia ja helpottaa samalla useiden molekyylien kuljetusta solukalvon poikki.ABCA-alaheimon 90 jäsentä ovat kaikki mukana lipidikuljetuksissa.91 puutteellinen ABCA12-toiminto aiheuttaa lipidikuljetushäiriöitä lamellaarisissa elimissä ja johtaa siten solujenvälisten lipiditasojen vähenemiseen marraskedessa.3ultrastrukturaaliset tutkimukset ovat osoittaneet, että ABCA12 sijaitsee lamellaarisissa kappaleissa, jotka liittyvät glykosyyliseramideihin.91abca12 mutaatiot ovat liittyneet glykosyyliseramidien jakautumisen ja kuljetuksen häiriöihin sekä hydroksikeramidien, joka on yksi solujen välisissä tiloissa olevan lipidiesteen pääkomponenteista, alentuneisiin pitoisuuksiin.3,6,92,93 näillä potilailla esiintyvä massiivinen hyperkeratoosi voi olla kompensoiva vaste puutteelliselle rasva-esteelle.94 se saattaa johtua myös sarveiskalvosyyttien hilseilyn puuttumisesta, 93, joka voi johtua tiettyjen proteaasien, kuten kallikreiini 5: n ja katepsiini D: n, kuljetusvirheistä, jotka johtuvat lamellikappaleiden häiriöistä.95 hiirimallia ja in vitro-tutkimukset viittaavat siihen, että ABCA12-mutaatioilla on vaikutusta myös epidermaaliseen erilaistumiseen.95-97
tähän mennessä ABCA12-geenissä on raportoitu yli 50 mutaatiota ARCI-potilailla Afrikasta, Euroopasta, Pakistanista ja Japanista. Yleisimmät mutaatiot ovat pakistanilaisissa ja intialaisissa populaatioissa tunnistettu p.Val244SerfsTer28,2,98,99 ja afrikkalaisissa perheissä tunnistettu p.Asn1380Ser, 4. Molemmissa tapauksissa nämä voivat olla perustavia mutaatioita.
ABCA12-mutaatioiden laajuus liittyy fenotyyppiin, ja täydelliseen toimintakyvyttömyyteen liittyvät mutaatiot johtavat HI-fenotyyppiin.2,3,98-102 sen sijaan LI: ssä ja CIE: ssä suurin osa mutaatioista on puutteellisia, ja niillä on lievempi vaikutus proteiinin toimintaan.4-6,103 LI-fenotyypin taustalla olevat mutaatiot näyttävät keskittyvän ensimmäiseen adenosiinitrifosfaattia sitovaan kasettialueeseen.4 kliinisesti potilailla, joilla on CIE ja mutaatioita ABCA12-geenissä, on keskikokoisia asteikkoja, jotka ovat jonkin verran suurempia kuin yleensä tätä fenotyyppiä sairastavilla potilailla.
Harlekiinisikiö
HI eli harlekiinisikiö on vakava ja yleensä kuolemaan johtava iktyoosin muoto. Lapset ovat yleensä ennenaikaisia laajoja kiiltäviä hyperkeratoottisia plakkeja, joita erottavat syvät halkeamat, jotka peittävät koko kokonaisuuden ja muodostavat geometrisia kuvioita, jotka muistuttavat harlekiinien käyttämiä vaatteita, jolloin ehto on saanut nimensä. Ihon kireys johtaa silmäluomien ja huulten huomattavaan eversioniin, nivel-ja nenäruston alkeelliseen kehittymiseen ja toisinaan mikrokefaliaan. Lapsilla on harvoin ripsiä tai kulmakarvoja, vaikka hiuspohjan karvat saattavat säilyä. Kädet ja jalat ovat turvonneet ja edematoiset, ja niitä peittää usein käsinettä muistuttava kerros. Niillä voi olla sormikuristuksia.
tällaisten potilaiden riski kuolla vastasyntyneen aikana on erittäin suuri.104 keuhkojen ilmanvaihto vaarantuu; transepidermaalinen vesihäviö johtaa nestehukkaan, vesivoimalan epätasapainoon ja lämpö epävakauteen; ja infektioiden riski kasvaa. Kasvojen kireys ja eklabium haittaavat imemistä ja siten ruokintaa, jolloin vastaava nestehukan paheneminen. Vastasyntyneet, joilla on tämä ehto, elivät harvoin pidempään muutamaa viikkoa. Viime vuosina mahdollisuudet pitkäaikaiseen eloonjäämiseen ovat kuitenkin lisääntyneet huomattavasti, mikä johtuu pääasiassa systeemisten retinoidien antamisesta ja vastasyntyneiden tehohoidon edistymisestä.105 tuoreessa tutkimuksessa 83% oraalisilla retinoideilla hoidetuista potilaista selvisi hengissä, kun hoitamattomista potilaista vastaava luku oli 24%. Suurin osa kuolemista tapahtui ensimmäisten 3 elinpäivän aikana, mutta hoito aloitettiin vasta tämän jälkeen monilla eloonjääneillä.Tämä viittaisi siihen, että monet näistä varhaisista kuolemista olisivat tapahtuneet retinoidihoidosta riippumatta.
vastasyntyneillä lapsilla kehittyy yleensä vaikea CIE.Abca12-geenin mutaatioiden luonne ja sijainti sekä kuljettajatoimintojen häviämisen laajuus voivat määrittää ennusteen.3,92,107 potilaalla, jotka säästävät tietynasteista proteiiniaktiivisuutta, vaikkakin vähäistä, voi olla paremmat mahdollisuudet selviytyä. Homotsygoottisten mutaatioiden kantajilla on suurempi kuolleisuus.104
HI: n tärkein histologinen ominaisuus on erittäin paksun ja tiiviin ortokeratoottisen marraskeden esiintyminen. Karvatupet ja hiki kanavat ovat näkyvästi hyperkeratoottinen plugs107, 108 ja on epänormaali tai puuttuu lamellaariset elimet, lipidi sulkeumat, tai jäänteitä organelles tai ytimiä sarveissyyttien, ja puuttuminen solujenvälisten lipidien ultraäänitutkimuksessa.108,109 karvatupet osoittavat huomattava pitoisuus keratoottista materiaalia, joka on diagnostinen ominaisuus HI käytetään synnytystä diagnoosi.
tähän mennessä ABCA12-geenin mutaatioiden toteamisnopeus HI-potilailla on lähes 100%, joten kyseessä näyttäisi olevan geneettisesti homogeeninen tila.
Kollodiumlapsi ja Itseparantuva Kollodiumlapsi
Kollodiumlapsi syntyy yleensä ennenaikaisesti ja perinataalinen sairastuvuus ja kuolleisuus lisääntyy. Syntyessään vastasyntynyttä peittää kiiltävän opettava läpinäkyvä kalvo, joka muistuttaa sellofaanikäärettä (viikuna. 5). Vauvoilla on ektropionia, eklabiumia sekä nenän ja nivelruston hypoplasia. Imeminen ja keuhkotuuletus saattavat estyä110, ja transepidermaalinen vedenhukka ja infektioriski kasvavat.110,111

Kollodionivauva, joka myöhemmin eteni lamellaariseen iktyoosi-fenotyyppiin.
Collodion baby on HI ja CIE: n tavanomainen esitys. Autosomaalinen dominantti LI, 112,113 Sjögren-Larssonin oireyhtymä,110 trikothyodystrofia,114 juvenile Gaucher ‘ n tauti, 110 neutraalin lipidivaraston sairaus, Conradi-Hünermann-Happlen oireyhtymä, Hays-Wellsin oireyhtymä ja ektodermaalinen dysplasia115 voivat myös esiintyä satunnaisesti kollodium-vauvana. Kalvo katoaa spontaanisti 10-24 prosentilla vastasyntyneistä, jolloin iho muuttuu täysin normaaliksi.110 116 aiemmin näitä tapauksia kuvattiin vastasyntyneen LI: ksi, 117: ksi, mutta niistä ei käytetä nimitystä SHCB.118 jotkut kirjoittajat ovat ehdottaneet termiä itsestään paraneva kollodion ichthyosis, koska monet näistä potilaista, kun tutkitaan uudelleen myöhemmin lapsuudessa tai aikuisina, on vaihteleva anhidrosis ja lämpö intoleranssi ja lieviä merkkejä iktyoosi, kuten xerosis ja hieno desquamation, erityisesti axillae ja kaulan.78
kollodium-vauvan Optiset mikroskopiat tai ultrarakenteiset tutkimukset eivät ole spesifisiä. Siksi on suositeltavaa lykätä ihobiopsiaa, kunnes lopullinen fenotyyppi on kehittynyt.
Shcb-potilailla on todettu mutaatioita TGM1 -, 7 -, 119ALOKSE3 -, 78-ja ALOX12B23 -, 78-ja 79-geeneissä. ALOX12B-mutaatiot ovat yleisimpiä. 15 pohjoismaalaisen SHCB-potilaan sarjassa 67%: lla oli mutaatioita ALOX12B-geenissä, 25%: lla ALOXE3-geenissä ja 8, 3%: lla TGM1-geenissä.78 mutaatiota ei löydetty kaikilta potilailta, joten myös muut geenit ovat todennäköisesti mukana. On spekuloitu, että nämä mutaatiot vähentävät entsymaattista aktiivisuutta kohdussa, mutta eivät syntymän jälkeen.7 kohdussa, jossa hydrostaattinen paine on suuri, veden Kelaatio muuttaa mutatoituneen entsyymin inaktiiviseksi konformaatioksi. Syntymän jälkeen paineen laskiessa entsyymi palaa aktiiviseen muotoonsa ja sen aktiivisuus kasvaa riittävästi normaalin tai minimaalisesti vaikuttavan fenotyypin ylläpitämiseksi.
Akraalinen itsestään paraneva Kollodium-vauva
vaikka kollodium-vauva vaikuttaa koko kehoon, on raportoitu tapauksia, jotka rajoittuvat akraalialueille. Vuonna 1952 Finlay et al.120 ilmoitti kollodium-kalvotapauksesta, joka vaikutti vain käsiin ja jalkoihin ja joka seurasi itsestään paranemista. Viime aikoina on raportoitu uudesta acral SHCB-tapauksesta, joka liittyy TGM1-geenin mutaatioihin.8 ei tiedetä, miksi nämä vauriot rajoittuvat akraalialueille, vaikka entsyymiaktiivisuuden paikkariippuvaiseen säätelyyn liittyvät tekijät saattavat olla toiminnassa.8
Bathing Suit Ichthyosis
Bathing suit ichthyosis raportoitiin ensimmäisen kerran itsenäisenä ARCI-muunnoksena vuonna 2005, vaikka erikoisen levinneisyyden omaavia iktyoositapauksia oli raportoitu jo aiemmin.121-123 sitä on havaittu pääasiassa eteläafrikkalaisilla potilailla, 9 vaikka sitä on raportoitu myös yksilöillä Euroopasta ja Välimeren maista.124 syntyessään potilailla on yleistynyt kollodium kalvo, joka sitten vuodat jättää tyypillinen jakautuminen skaalaus. Runko, proksimaalinen alue käsivarsien, kuten axillae, kaula, ja päänahka ovat yleensä vaikuttaa, kun taas keskiosa kasvot, raajat, ja lisämunuaisen alue ovat yleensä säästynyt.9 suomut ovat suuria, lamellimaisia ja väriltään tummia. Hienompaa desquamaatiota voi esiintyä popliteaalisessa ja antekubitaalisessa fosseassa.124125 kämmenissä ja jalkapohjissa on lievä diffuusi hyperkeratoosi, kun taas kämmenissä ja jaloissa ei näy mitään.
sairaan ihon Histopatologisessa tutkimuksessa havaittiin huomattava hyperkeratoosi ilman aurinkokeratoosia, normaalit rakeiset kerrokset, lievä tai kohtalainen akantoosi ja lievä lymfosyyttinen infiltraatti ylänahkassa.9 Elektronimikroskopiahavainnot vastaavat useimmissa tapauksissa synnynnäistä tyypin 2 iktyoosia. Sekaantumaton iho ei näytä mitään epänormaaleja löydöksiä.124 125 terveellä iholla TGase 1: n aktiivisuus on hieman vähentynyt ja se on yleensä paikallistunut perisellulaarisille alueille. Mukana iho, entsymaattinen aktiivisuus on jäljellä ja epänormaalisti sijaitsee sytoplasma.124
TGM1-geenissä on havaittu mutaatioita kaikilla tähän mennessä tutkituilla uimahousujen iktyoosia sairastavilla potilailla.119,124-126 yleisin mutaatio on P.Arg315Leu, joka on tunnistettu useimmilla eteläafrikkalaisilla potilailla ja voi olla perustajamutaatio. Oji ym.124 ehdotti, että ihon lämpötilalla voisi olla osuutta näiden ilmenemismuotojen kehittymiseen. Digitaalisen termografian avulla tekijät osoittivat vahvan korrelaation ruumiinlämmön ja desquamaation välillä, sillä kaikkein kuumimmat kehon alueet kärsivät eniten. Aufenvenne ym.127: ssä todettiin TGase 1: n aktiivisuuden optimilämpötilan lasku uimahousupotilailla, joilla oli iktyoosi. Tätä laskua ei havaittu terveillä verrokeilla eikä potilailla, joilla oli yleistynyt LI. tämä lämpötilan lasku selittäisi näiden potilaiden fenotyypin. Normaalille entsyymille optimaalinen lämpötila on 37°C mutta mutatoituneelle entsyymille 31°C.
hoito
hoidon ensisijainen tavoite iktyoosissa on poistaa skaalaus ja vähentää kseroosia aiheuttamatta liiallista ärsytystä (Taulukko 3). Ennen kuin päätetään hoidosta, on otettava huomioon muun muassa potilaan ikä ja sukupuoli, taudin tyyppi ja vakavuus sekä vaurioiden laajuus ja sijainti.128
terapeuttinen strategia Autosomaalisessa Resessiivisessä synnynnäisessä Iktyoosissa.
| terapeuttinen strategia autosomaalinen resessiivinen synnynnäinen ichthyoses | |
| uiminen ja vaakojen mekaaninen poistaminen | uiminen natriumbikarbonaatilla, vehnätärkkelyksellä, maissitärkkelyksellä tai riisitärkkelyksellä; vaa ‘ an mekaaninen poisto (1 tai 2 kertaa päivässä) |
| paikallishoito (juokseva) | ureaa sisältävät kosteusvoiteita sisältävät keratinolyytit propyleeniglykolilla yhdistetyin keratinolyytein (propyleeniglykoli, α-hydroksihapot tai urea)Keratinolyytit yhdistettynä salisyylihappoontopisiin retinoideihin vastasyntyneillä ja pienillä lapsilla, sovelletaan kantaja-ainetta ilman vaikuttavia aineita. Vältä ureaa, salisyylihappoa ja maitohappoa systeemisen imeytymisriskin vuoksi |
| oraalinen hoito | oraaliset retinoidit (asitretiini tai isotretinoiini) |
| muut toimenpiteet | ektropionin seuranta oftalmologisella ulkokorvan säännöllisellä puhdistuksella korva-kurkku-nenä-erikoisfysioterapialla supistusten ehkäisemiseksi.Rasittavien toimintojen välttäminen korkeassa ympäristön lämpötilassahydroterapia |
päivittäinen uiminen ja vaakojen mekaaninen poistaminen
suositellaan ARCI-potilaille, jotta vaa ‘ at ja kosteusvoiteen jäämät voidaan mekaanisesti poistaa. Tämä on helpompaa, jos potilas upotetaan veteen 15-30 minuuttia. Jotkut kirjoittajat suosittelevat natriumbikarbonaatin lisäämistä kylpyyn keratiinien denaturalisoimiseksi ja veden tekemiseksi emäksiseksi ja siten helpottamaan asteikkojen poistamista.129 muita tuotteita, joita voidaan lisätä, ovat vehnätärkkelys, maissitärkkelys tai riisitärkkelys. Uimaöljyt eivät ole tarkoituksenmukaisia, koska ne voivat aiheuttaa tukoksen, josta seuraa bakteerien lisääntymisen ja lämmönsäätelyn pahenemisen vaara.
paikallishoito
kosteusvoiteet ja keratolyyttiset aineet ovat yleensä ensimmäinen hoitovaihtoehto. Ne parantavat ihoesteen toimintaa ja helpottavat hilseilyä. Lieviä paikallisia haittavaikutuksia, kuten ohimenevää kutinaa, ärsytystä tai kirvelyä saattaa esiintyä.
natriumkloridia, ureaa, E-vitamiiniasetaattia, glyserolia ja vaseliinia voidaan käyttää kosteusvoiteina ja voiteluaineina. Potilailla, joilla on paksu skaalaus ja huomattava hyperkeratoosi, voidaan lisätä 1 tai useampia keratolyyttisiä aineita,kuten α-hydroksihappoja (maitohappo ja glykolihappo), 130 salisyylihappoa,N-asetyylikysteiiniä,131-133 ureaa (>5%), 134 ja propyleeniglykolia. Myös keratinosyyttien erilaistumisen modulaattoreita käytetään. Näitä ovat paikallisesti käytettävät retinoidit (tretinoiini, adapaleeni,tatsaroteeni), 135,136 kalsipotrioli, 137 ja dekspantenoli.Ajankohtaiset retinoidit aiheuttavat usein ärsytystä ja pieniä, hyvin kivuliaita halkeamia.Lisäksi hedelmällisessä iässä olevilla naisilla on imeytymisen ja teratogeenisuuden riski, jos niitä käytetään liian paljon.138 keratolyyttien ja kosteusvoiteiden tehokkuuden parantamiseksi okklusiivista sidosta voidaan soveltaa tietyillä alueilla, jotka ovat tulenkestäviä hoitoon.139 lisäaine tai synergistinen vaikutus voidaan saavuttaa myös yhdistämällä 2 tai useampia keratolyyttisiä aineita tai kosteusvoiteita.140-142 hoito on optimoitava jokaiselle yksilölle, koska tilan luonne ja ihon herkkyys vaihtelevat suuresti ja kunkin hoidon vasteessa on eroja. Optimointiprosessia voidaan auttaa käsittelemällä kehon toista puolta eri tavalla, jotta vertailu olisi mahdollista. Vastasyntyneitä ja pieniä lapsia tulisi hoitaa ajoneuvolla, jossa ei ole vaikuttavia aineita, koska iho on erittäin hieno ja herkkä ja useimmat keratolyytit eivät siedä. Lisäksi paikallisten valmisteiden, kuten urean, salisyylihapon ja maitohapon, perkutaanisen imeytymisen riski on suurempi.143-145
systeeminen hoito
suun retinoideilla on keratolyyttisiä vaikutuksia, jotka auttavat poistamaan asteikkoja ja ehkäisemään liiallista hyperkeratoosia. Sekä isotretinoiini että aromaattiset retinoidit (asitretiini ja etretinaatti) ovat osoittautuneet tehokkaiksi Arciksen hoidossa.128 146 147 Asitretin annos 0,5-1 mg/kg/d on yleisimmin käytetty lääke, erityisesti LI-potilailla.148 CIE-potilaalla saattaa olla täydellisempi vaste ja pienemmät annokset.
tärkeimmät haittavaikutukset ovat mukokutaaniset häiriöt, teratogeenisuus, tuki-ja liikuntaelinten sairaudet sekä epänormaali lipidiprofiili ja kohonneet transaminaasiarvot.149-152 teratogeenisuuden osalta etretinaatin ja asitretiinin käyttöä tulee välttää raskauden aikana ja potilaiden tulee välttää raskaaksi tulemista 3 vuoden ajan hoidon lopettamisen jälkeen.151 Isotretinoiinilla on lyhyempi puoliintumisaika ja se eliminoituu kokonaan elimistöstä 1 kuukauden kuluttua, joten se voi olla ensisijainen vaihtoehto naisille, jotka haluavat tulla raskaaksi.128
hoidon seurantaan tulee kuulua laboratoriokoe, jossa tehdään maksan toimintakoe ja lipidiprofiili ennen hoidon aloittamista, sitten 1 kuukauden kuluttua ja 3 kuukauden välein hoidon aloittamisesta. Hedelmällisessä iässä oleville naisille raskaustesti on tehtävä 2 viikkoa ennen hoidon aloittamista ja tehokasta ehkäisymenetelmää on käytettävä 4 viikkoa ennen hoitoa ja 3 vuotta sen jälkeen (asitretiinin tapauksessa). Jos retinoidihoito on pitkittynyttä, kasvua ja luun kehitystä on seurattava. Jotkut kirjoittajat ehdottavat luustotutkimuksen tekemistä ennen hoitoa, jota seuraa vuosittainen tutkimus.151 Viimeaikaiset ohjeet eivät suosittele rutiiniradiografian suorittamista mahdollisten haittavaikutusten vuoksi.Sen sijaan suositellaan selektiivisiä röntgentutkimuksia potilaille, joilla on epätyypillistä luukipua.
vaihtoehto systeemiselle retinoidihoidolle on käyttää retinoiinihapon metabolian estäjiksi kutsuttuja lääkkeitä, jotka lisäävät endogeenisen retinoiinihapon pitoisuutta. Yksi tällainen lääke on liarotsoli, jolle Euroopan lääkevirasto ja Yhdysvaltain elintarvike-ja lääkevirasto ovat myöntäneet orvon aseman LI -, CIE-ja HI-virusten hoidossa.153-155 tämä lääke on osoittautunut tehokkaammaksi kuin acitretiini kliinisissä tutkimuksissa ja se on myös paremmin siedetty ja sillä on parempi farmakokineettinen profiili.154
muu lääketieteellinen hoito
ektropionipotilailla keinotekoisten kyynelten ja silmän voiteluaineiden käyttö sekä kasvojen ja erityisesti poskien ihon kosteuttaminen voivat vähentää palpebraalista vetäytymistä. Kirurginen korjaus on varteenotettava vaihtoehto vaikeissa tapauksissa, mutta tämä on yleensä toistettava muutaman vuoden kuluttua. Vesiterapiasta voi olla hyötyä.Potilaita tulee neuvoa välttämään rasittavaa fyysistä aktiivisuutta, kun ympäristön lämpötila on korkea, koska hypohidroosiin liittyy lämpöhalvauksen ja kouristusten vaara. Suun retinoidit voivat parantaa lämmönsäätelyä.157 fysioterapia on tärkeää fleksion kontraktuuran ehkäisyssä, erityisesti HI-taudin yhteydessä. Korva-kurkku-nenä-erikoislääkärin suorittama ulkoisen korvakäytävän säännöllinen puhdistus voi estää suomujen kertymisen ja siten estää kuulon heikkenemisen.
geneettinen neuvonta ja synnytystä edeltävä diagnoosi
kun potilaalla todetaan iktyoosi, hänelle tulee tarjota asianmukaista geneettistä neuvontaa, jossa selvitetään häiriön luonne, tartuntatapa ja tulevien oireiden riski perheessä. Raskausdiagnoosi voi osoittaa, vaikuttaako sikiö, ja jos näin on, voidaan tarjota perheen psykologista valmistelua ja ennakoida ongelmia raskauden ja synnytyksen aikana. Vanhemmille voidaan antaa mahdollisuus aborttiin, jos hoitoa ei ole saatavilla. Lisäksi, jos geeniterapiaa näitä ehtoja tulee saataville tulevaisuudessa, synnytystä diagnoosi mahdollistaisi soveltamisen tämän hoidon mahdollisimman varhaisessa vaiheessa.
yli 20 vuoden ajan sikiödiagnoosi tehtiin ottamalla sikiön ihosta biopsianäyte ja tutkimalla sitä optisella mikroskopialla, elektronimikroskopialla tai immunohistokemialla.158 159 tämä invasiivinen toimenpide voitiin tehdä vain raskauden loppuvaiheessa, raskausviikkojen 15 ja 23 välillä, ja siihen liittyi 1-3 prosentin riski menettää sikiö.160 161 perinnöllisten ihosairauksien molekyylimekanismien tunnistaminen on mahdollistanut paljon aikaisemman diagnoosin, joka perustuu geneettisiin tekniikoihin.102 162–164 sikiön DNA saadaan amniocentesis suoritetaan välillä viikoilla 15 ja 20 tai korioninen villus näytteenotto välillä viikoilla 10 ja 12. Sikiön menetyksen riski näillä tekniikoilla on alle 0,5 – 1%.165 muita kehitteillä olevia noninvasive-menetelmiä ovat sikiön solujen DNA: n ja vapaan sikiön DNA: n analysointi äidin kiertokulussa166 sekä 3-ulotteisen ultraäänen käyttö.167,168
preimplantaatiota edeltävä geneettinen diagnoosi voisi olla mahdollinen myös koeputkihedelmöitysmenetelmissä siten, että kohtuun istutetaan vain hedelmöittyneitä munasoluja, joissa ei ole mutaatiota, jolloin useimmissa tapauksissa ei tarvita aborttia.169
tulevat strategiat iktyoosin geneettistä hoitoa varten
vaikka iktyoosin geneettisessä diagnosoinnissa on edistytty merkittävästi, uusia strategioita jatketaan myös näiden sairauksien osalta.170 iho on helpoin elin geeninsiirtohoidoissa, joten tällaiset tekniikat ovat minimaalisesti invasiivisia.171 iholla on kuitenkin myös ainutlaatuisia immunologisia ominaisuuksia, jotka eivät suosi siirtogeenisen tuotteen pitkäaikaista ilmentymistä.172 LI: ssä ex vivo-geeninsiirtoprosessi onnistui palauttamaan normaalin TGM1-ilmentymän ja korjaamaan immunosuppressiivisten hiirten selkään siirretyn ihon fenotyypin.173 174 äskettäin on myös löydetty abca12-geenin mutaatioiden takia HI-potilailla viljeltyjen keratinosyyttien fenotyyppi.3
eturistiriidat
kirjoittajat ilmoittavat, ettei heillä ole eturistiriitoja.