Frontiers in Neurology
Introduction
Neuroacanthocytosis is a superordinate term for a group of rare syndromes that are character by neurological oires in combination with piikikäs deformated red blood cells (acanthocytes). Tämä raportti keskittyy Chorea-acanthocytosis (ChAc) , joka on orpo sairaus arviolta 1,000 vaikuttaa yksilöiden maailmanlaajuisesti aiheuttama autosomaalinen-resessiivinen mutaatioita vps13a (vacuolar protein lajittelu 13 homolog A) geeni kromosomissa 9q (1). Tauti kulkee etenevä, syitä lisääntynyt kuolleisuus voi olla vähentynyt motoriset toiminnot korreloivat riskitekijöitä, kuten dysfagia, mutta myös äkillisiä odottamattomia kuolemia on kuvattu (2, 3). Toistaiseksi ei ole olemassa mitään aiheuttavaa hoitoa.
asantosytoosin ja liikehäiriöiden epäilyttävästä rinnakkaiselosta raportoivat ensimmäisen kerran Levine ja Critchley (4, 5) 1970-luvulla, ja se on sittemmin hyväksytty taudin huomattavaksi piirteeksi. Kuitenkin meidän tapausraportti kuvataan harvinainen kliininen fenotyyppi ChAc puuttuu ilmeinen liikkeen häiriöt, mikä viittaa laajempaan erilaisia kliinisiä esityksiä. Diagnostisten työkalujen on vastattava näihin haasteisiin oikean diagnoosin määrittämiseksi. ehdotamme tässä molekyylin neuroimagingia keskeiseksi menetelmäksi.
Tapausraportti
miesindeksipotilas sai ensimmäisen kerran 25-vuotiaana kaksi provosoimatonta molemminpuolista toonis-kloonista kohtausta. Sairauskertomusta ei ollut. Aivojen magneettikuvaus ja aivosähkökäyrä olivat tuossa iässä normaalit, eikä hoitoa aloitettu potilaan haluttomuuden ja harvojen tapahtumien vuoksi. Potilaalla oli kuitenkin jatkuvia kohtauksia, jotka nyt esiintyvät osittaisena epilepsiana. Hän kuvaili toistuvan äkillisen huimauksen tunteita, jotka diagnosoitiin vertigiiniseksi auraksi. Lisäksi hänellä oli dyskognitiivisia kohtauksia, joissa hän osoitti joko A) reagoimattomuutta ja turkkilaisten rukousten lausumista, b) suullisia automatismeja tai c) Käsien näpelöintiä ja ääntelyä—kaikki osittain kehittymässä toonis-kloonisiksi kohtauksiksi. Video-EEG: ssä näkyi nyt paikallisia piikki – aaltokomplekseja sekä oikeassa että vasemmassa ohimolohkossa. Kouristuskohtauksen semiologian mukaisesti tehtiin diagnoosi molemminpuolisesta ohimolohkoepilepsiasta ja aloitettiin lääkitys levetirasetaamilla.
31-vuotiaana kohtauksia ei täysin tukahdutettu, ja osana ohimolohkoepilepsian jatkodiagnostiikkaa potilaalle tehtiin FDG-PET (Kuva 1), jossa ilmeni molemminpuolista mesiotemporaalista hypometaboliaa mesiotemporaalisen kohtauksen alkuperän mukaisesti. Kuitenkin oli myös huomattava, erittäin epätavallinen striataalinen hypometabolismi, joka herätti epäilyn hermoston rappeutumissairaudesta. Tämän vuoksi käynnistettiin laaja seuranta-arviointi (KS.Kuvio 2). FP-CIT-SPECT tehtiin, jolloin striataalisen dopamiinin kuljettajaproteiinin saatavuus väheni kohtalaisesti molemminpuolisesti, mikä oli linjassa nigrostriataalisen eheyden heikkenemisen kanssa (kuva 1). Korkean resoluution magneettikuvauksessa todettiin nyt tuman caudatus molemminpuolinen atrofia (kuva 3). Neurologisissa tutkimuksissa havaittiin erillinen sidottu kävelykuvio ja hypomimia, mutta ei muuta ekstrapyramidaalimotoriikan kiintymystä eikä bulbaarioireita, kuten ruokintadystoniaa. Lukuun ottamatta alaraajojen vähäistä refleksitilaa, jossa hermojen johtumistutkimukset vahvistivat sensorimotorisen aksonaalisen polyneuropatian, neurologinen tutkimus oli normaali. Neuropsykologisiin oireisiin kuului muun muassa ahdistuneisuushäiriön piirteitä, ja potilaalla todettiin vaikeuksia lähimuistin heikkenemisessä. Neuropsykologiset testit eivät kuitenkaan vahvistaneet ilmeistä mnestistä toimintahäiriötä, vaan pikemminkin epäspesifistä häiriötekijää, jota saattoi lisätä myös siihen aikaan puutteellinen kohtausten tukahduttaminen. Laboratoriokokeet olivat negatiivisia minkä tahansa oireisen epilepsian (ts., autoimmuuni-enkefaliittivasta-aineet, antineuronaaliset vasta-aineet). Aivo-selkäydinnesteessä ei havaittu tulehduksen merkkejä, mutta proteiinipitoisuus suureni kohtalaisesti (765 mg/l). Biogeenisten amiinien analyysi aivo-selkäydinnesteestä paljasti kohonneen glutamiinin määrän, jonka yhdistimme viimeaikaisiin kohtauksiin. Ääreisveressä akantosyyttejä oli 0,4%. Wilsonin taudin serologinen testaus oli negatiivinen. Huomattavaa oli jatkuva kreatiinikinaasin nousu (vaihteluväli: 1 000–3 000 U/l), joka johti epäiltyyn mitokondrion häiriön diagnoosiin. Lisätutkimuksia varten tehtiin iskeeminen laktaattitesti, jonka tulokset olivat normaalit. M. gastrocnemicuksen lihasbiopsia tehtiin, mutta mitokondrioiden toiminnan ja hengitystieketjun analyysi oli normaali.
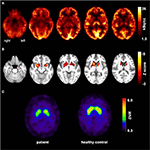
Kuva 1. Molekyylikuvaustutkimusten tulokset. Transaksiaaliset FDG-PET-kuvat (a) osoittivat paitsi lievää kahdenkeskistä mesiotemporaalista hypometaboliaa (mesiotemporaalisen kohtauksen alkuperän mukaisesti), myös huomattavaa striataalista hypometaboliaa, joka todettiin erittäin merkittäväksi verrattuna terveisiin kontrolleihin . Lisäksi tehdyssä FP-CIT-SPECT-tutkimuksessa (C) havaittiin myös striataalisten dopamiinikuljettajien Kohtalaista häviämistä, todistaen nigrostriataalisen eheyden heikkenemistä (Ikä-matched terve kontrolli on esitetty vertailua varten; DVR, jakelutilavuussuhde).

kuva 2. Index-potilaan kliininen ja diagnostinen kurssi.
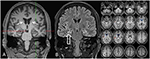
kuva 3. Magneettikuvauksessa näkyy diskreetti molemminpuolinen pään atrofia ja pienempi ja hyperintensoitunut oikeanpuoleinen hippokampus, joka viittaa oikeanpuoleiseen hippokampuksen skleroosiin . Vasen hippokampus on hieman hyperintense, mutta ei atrofinen. Molemminpuolinen caudate head atrofia vahvistetaan CVR-analyysillä, jossa sininen väri osoittaa vähentynyt harmaa ja punainen väri lisääntynyt aivo-selkäydinnesteen tilavuus vastaavasti (C).
sukuhistoria paljasti hänen vanhempiensa (Ensimmäiset serkut) verisukulaisuuden, joilla itsellään ei ollut merkittävää sairaushistoriaa. Hänen kuudesta sisaruksestaan kaksi (ikä 20-30 vuotta) oli tähän mennessä kärsinyt myös yleisistä kohtauksista (KS.Kuva 4). Hänen sisarustensa potilastiedot eivät olleet saatavilla.
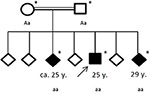
Kuva 4. Sairastuneen perheen sukutaulu, jonka ikä on ensimmäinen epileptinen kohtaus vuosina (y). Nuoli: indeksipatentti. Asteriski: geneettinen testaus suoritettu. Aa, heterotsygootti; aa, homotsygootti C.4326 t>A (S.Tyr1442*).
perinnöllisen neurodegeneratiivisen liikehäiriön hypoteesin perusteella potilas ja hänen perheensä lähetettiin geneettisiin testeihin. Exome-sekvensointi paljasti homotsygoottisen romaanin typistävän mutaation c. 4326 t>A (P.Tyr1442*) Chac-geenissä VPS13A kolmessa sairastuneessa sisaruksessa. Molemmat konsangiinivanhemmat todettiin heterotsygooteiksi. Exome-sekvensoinnissa ei havaittu muita variantteja tai mutaatioita.
itse asiassa 33-vuotiaana tehdyssä seurannassa indeksipotilaalla oli edelleen vain marginaalisia ekstrapyramidaalioireita, eikä yhdelläkään hänen sisaristaan esiintynyt mitään. Nyt hän antoi vaikutelman lievästä jäykkyydestä vain oikean yläraajan pohjustuksessa ja lievästä yläraajojen bradykinesiasta. Levetirasetaami-lakosamidihoidossa potilaalla ei ole esiintynyt kouristuskohtauksia.
Keskustelu
tässä tapauksessa osoitamme, että ChAc voi kliinisesti ilmetä ohimolohkoepilepsialla, josta puuttuu merkittäviä motorisia oireita, jotka johtuvat uudesta vastaavasta mutaatiosta VPS13A-geenissä.
taudinkulkua luonnehtivat tyypillisesti etenevä liikehäiriö (mm.Koreaa, dystoniaa, parkinsonismia), kognitiiviset ja psykiatriset muutokset sekä myopaattiset oireet, joihin liittyy serologisia merkkejä akantosytoosista ja hyperckemiasta. Kouristuskohtaukset on aiemmin todettu ChAc: n (6) oireeksi, mutta liikehäiriöitä pidetään edelleen keskeisenä oireena. On todisteita siitä, että epilepsia, ja tarkemmin ottaen ohimolohkosta peräisin oleva epilepsia, saattaa olla aliarvioitu Chac: n fenotyyppi. Peluso ym. raportoitu potilas samanlainen kuin meidän, joka, edes 46 vuoden iässä, ei osoittanut liikehäiriöitä, kun neuroimaging tulokset viittaavat tyvitumakkeiden osallisuudesta (7). Tämän mukaisesti, Scheid et al. kuvattu kolme potilasta, joilla oli mesiaalinen ohimolohkoepilepsia ja skleroosi (MAGNEETTIKUVAUSTUTKIMUSTEN perusteella) vallitsevana oireena ja joilla diagnosoitiin ChAc (8). Lisäksi Mente et al. äskettäin toimitettu ruumiinavaus tulokset Chac potilas, joka osoitti paitsi tyvitumakkeiden surkastumista, mutta myös hippokampaaliskleroosi (9). Meidän tapauksemme mukaisesti hippokampuksen kahdenvälinen osallisuus näyttää olevan yleinen havainto (10, 11). Tarkka yhteys kohtausten ja skleroosin välillä on kuitenkin edelleen kysymys kanasta tai munasta. Koreiinin korreloivaa roolia hippokampuksen rakenteellisessa kehityksessä ei vielä täysin ymmärretä. Mielenkiintoista on, että hiirimallissa Chac: n rakennemuutos nähtiin, kun GABA (a) – reseptorin gamma 2: n ja sen ankkuriproteiinin koreiinin hippokampaalinen ilmentymä lisääntyi (12).
kliinisessä rutiinissa tämä fenotyyppi voi edelleen olla haastava oikean diagnoosin löytämisessä. Ensimmäinen ratkaiseva vihje kohti neurodegeneratiivista sairautta potilaallamme oli merkittävä lasku striataalisessa glukoosiaineenvaihdunnassa ja kohtalainen lasku nigrostriataalisessa eheydessä FDG-PET: llä ja FP-CIT-SPECTILLÄ. Pieni kokoelma toiminnallisia kuvantamistuloksia, jotka perustuvat tapaussarjan kuvaukseen, osoittaa, että muutoksia aineenvaihdunnassa ja dopaminergisiä häiriöitä esiintyy useimmiten caudate tumassa ja putamenissa (13). Aivojen magneettikuvaus paljasti Tuma caudatuksen atrofian sairauden aikana, mitä on kuvattu tyypilliseksi löydökseksi ChAc: ssa (14). Niinpä erillisiä neurologisia piirteitä ovat yleensä koreaia, ruokintadystonia, orofaciolingual dyskinesiat ja tics muiden tyvitumakkeiden kiintymyksen oireiden, kuten parkinsonismin ja dystonian (3) ohella. On houkuttelevaa spekuloida, että havaitut striataaliset muutokset potilaallamme olivat edelleen alle kynnyksen aiheuttaa oireita (eli prodromaalisia kuvantamislöydöksiä), jotka voivat lopulta tapahtua sairauden edetessä. Siksi kannustamme rakenne-ja molekyylikuvausta herkkänä diagnostisena välineenä tässä harvinaisessa sairaudessa.
Chac: ssa on kuvattu akantosyyttien määrän olevan 5-50% (3), mutta on olemassa muutamia tapausraportteja, jotka osoittavat, että akantosyyttien määrä saattaa ilmaantua vasta taudin loppupuolella (15), saattaa olla hyvin alhainen tai jopa puuttuu kokonaan (16). Siksi akantosyyttejä ei tule pitää pakollisina diagnoosin tekemisessä. Lisäksi epilepsia voi viivästyttää diagnoosi, koska se peittää muita tyypillisiä oireita ChAc. Erityisesti psykiatriset oireet, kuten persoonallisuuden muutokset ja kognitiivisten kykyjen heikkeneminen, jotka ovat hyvin yleisiä, voidaan tulkita väärin ja katsoa lääkkeisiin liittyviksi sivuvaikutuksiksi (levetirasetaami) tai kohtauksiin liittyviksi. Sekundaarinen hyperCKemia nähdään yleisten toonisten kloonisten kohtausten jälkeen ja jatkuva nousu saattaa jäädä huomaamatta.
jutussamme korostetaan myös eksome-sekvensoinnin ratkaisevaa hyötyä oikean diagnoosin aikaansaamiseksi, erityisesti harvinaisissa sairauksissa tai silloin, kun oireet ovat epämääräisiä. Nisäkäsperhe vps13 (A–D) – geenit kiinnostavat yhä enemmän, ja mutaatioita on havaittu muissakin neurologisissa sairauksissa, kuten autosomaalisessa resessiivisessä Parkinsonin taudissa tai autosomaalisessa resessiivisessä spinocerebellar ataksiassa (17). Resessiiviset mutaatiot vps13d: ssä aiheuttavat lapsuudessa alkavia liikehäiriöitä (18). Chac: n taustalla olevaa patofysiologiaa ei ole vielä täysin selvitetty, mutta osoitettiin, että VPS13A koodaa proteiinia, joka ilmenee kaikkialla aivoissa ja lukuisissa muissa kudoksissa (19). Tutkimukset osoittavat, että se osallistuu signalointireitteihin, jotka säätelevät sytoskeletaalista arkkitehtuuria, eksosytoosia ja solujen selviytymistä (20). Toinen mutaatio, jonka on kuvattu esiintyvän kohtausvaltaisella fenotyypillä 9 Chac-potilaalla, on C.2343del-mutaatio VPS13A-geenissä (21). On kohtuullista olettaa, että tietyt mutaatiot samassa geenissä aiheuttavat tietyn poikkeaman koreiinille, joka aiheuttaa ainutlaatuisen fenotyypin esimerkiksi vaikuttamalla aivojen eri alueisiin. Taustalla oleva patofysiologia on kuitenkin vielä vain spekulatiivista ja aiheuttavien mutaatioiden tarkempi dokumentointi kiinnostaa. Osoitamme tässä, että katkaiseva mutaatio c.4326 t>A (P.Tyr1442*), jota ei ole aiemmin kuvattu, johtaa samanlaiseen fenotyyppiin, joka on vallitseva epilepsia kaikilla sairastuneilla perheenjäsenillä.
Loppuhuomautukset
epilepsia (erityisesti molemminpuolinen ohimolohkoepilepsia) on todennäköisesti Chac: n vallitseva fenotyyppi. Siksi on huolellisesti etsittävä lisää punaisia lippuja (kuten hyperCKemia, sukuhistoria, polyneuropatia). Epäilyttävissä tapauksissa akantosyyttejä on etsittävä verestä ja aivoista, koska näitä välineitä on laajalti saatavilla. Epäselvissä tapauksissa diagnostiikkaa on täydennettävä FDG-PET-ja/tai FP-CIT-SPECT-valmisteilla, koska ne ovat herkkiä havaitsemaan striataalisen vaikutuksen. Autosomissa resessiivisesti periytyvissä epileptikoissa, joissa on merkkejä hermoston rappeumasairaudesta VPS13A, tulisi käyttää yhtä geenitestiä tai Koreiinia Western blotia oikean diagnoosin määrittämiseksi. Viimeksi mainittu tulisi ottaa huomioon myös muissa neurodegeneratiivisissa häiriöissä, joissa ei ole geneettisesti määriteltyä etiologiaa.
eettinen lausunto
potilaalta saatiin kirjallinen tietoon perustuva suostumus tutkimukseen osallistumiseen. Potilaalta saatiin kirjallinen tietoon perustuva suostumus tämän tapauskertomuksen julkaisemiseen.
Tekijäosuudet
JW, MR ja SK osallistuivat potilaiden hoitoon. JW kirjoitti käsikirjoituksen ensimmäisen luonnoksen. LF, HU, ja PM valmistettu lukuja. Kaikki kirjoittajat osallistuivat käsikirjoituksen tarkistamiseen, lukivat ja hyväksyivät toimitetun version.
Eturistiriitaselvitys
HU on osakas Veobrain Gmbh: ssa, joka on Freiburgin yliopiston lääketieteellisen keskuksen spin-off.
muut kirjoittajat ilmoittavat, että tutkimus tehtiin ilman kaupallisia tai taloudellisia suhteita, joita voitaisiin pitää mahdollisena eturistiriitana.
1. Walterfang M, Evans A, Leong Looi JC, Jung HH, Danek a, Walker RH, et al. Neuroakantosytoosioireyhtymien neuropsykiatria. Neurosci Biobehav Rev. (2011) 35:1275-83. doi: 10.1016 / J.neubiorev.2011.01.001
PubMed Abstract | CrossRef Full Text | Google Scholar
2. Walker RH. Neuroakantosytoosioireyhtymien hoito. Vapina Toinen Hyperkinetti. Mov. (2015) 5:346. doi: 10.7916 / D8W66K48
PubMed Abstract / CrossRef Full Text | Google Scholar
3. Velayos Baeza A, Dobson-Stone C, Rampoldi L, Bader B, Walker RH, Danek a ym. Koreaakantosytoosi. GeneReviews®. Seattle, WA: University of Washington (1993).
4. Critchley EM, Clark DB, Wikler A. Akantosytoosi ja neurologinen häiriö ilman Betalipoproteinemiaa. Arch Neurol. (1968) 18:134–40.
PubMed Abstract
5. Levine IM, Estes JW, Looney JM. Perinnöllinen neurologinen sairaus, johon liittyy akantosytoosi. Uusi syndrooma. Arch Neurol. (1968) 19:403–9.
PubMed Abstract / Google Scholar
6. Hardie RJ, Pullon HW, Harding AE, Owen JS, Pires M, Daniels GL, et al. Neuroakantosytoosi. kliininen, hematologinen ja patologinen tutkimus, jossa oli 19 tapausta. Brain (1991) 114(Pt 1a): 13-49.
PubMed Abstract / Google Scholar
7. Peluso S, Bilo L, Esposito M, Antenora A, Rosa ADR, Pappata s, et al. Chorea-acanthocytosis without chorea: laajentaa kliinistä fenotyyppiä. Parkinson Relat Disord. (2017) 41:124–6. doi: 10.2016 / J.parkreldis.2017.05.013
PubMed Abstract | CrossRef Full Text | Google Scholar
8. Scheid R, Bader B, Ott DV, Merkenschlager A, Danek A. mesiaalisen ohimolohkoepilepsian kehittyminen koreaakantosytoosissa. Neurology (2009) 73: 1419-22. doi: 10.1212 / WNL.0b013e3181bd80d4
PubMed Abstract | CrossRef Full Text | Google Scholar
9. Menne K, Kim SA, Grunseich C, Hefti MM, Crary JF, Danek A, et al. Hippokampaaliskleroosi ja mesiaalinen ohimolohkoepilepsia koreaacanthocytosis: tapaus kliinisellä, patologisella ja geneettisellä arvioinnilla. Neuropatoli Appl Neurobioli. (2017) 43:542–6. doi: 10.1111 / nan.12403
PubMed Abstract | CrossRef Full Text | Google Scholar
10. Bader B, Vollmar C, Ackl N, Ebert a, la Fougère C, Noachtar s, et al. Molemminpuolinen ohimolohkoepilepsia vahvistettu kallonsisäisellä EEG: llä koreaakantosytoosissa. Takavarikko (2011) 20:340-2. doi: 10.1016 / J.takavarikko.2010.12.007
PubMed Abstract | CrossRef Full Text | Google Scholar
11. Marson AM, Bucciantini E, Gentile E, Geda C. Neuroacanthocytosis: clinical, radiological, and neurofysiological findings in an Italian family. Neurol Sci. (2003) 24:188–9. doi: 10.1007 / s10072-003-0123-1
PubMed Abstrakti / CrossRef kokoteksti / Google Scholar
12. Kurano Y, Nakamura M, Ichiba M, Matsuda M, Mizuno E, Kato m, et al. Koreiinin puutos johtaa gephyriinin ja GABA(a) – reseptorin säätelyyn. Biochem Biophys Res Commun. (2006) 351:438–42. doi: 10.1016 / j. bbrc.2006.10.070
PubMed Abstract | CrossRef Full Text | Google Scholar
13. Ehrlich DJ, Walker RH. Functional neuroimaging and chorea: a systematic review. J Clin Mov Disord. (2017) 4:8. doi: 10.1186 / s40734-017-0056-0
PubMed Abstrakti / CrossRef kokoteksti / Google Scholar
14. Connolly BS, Hazrati L-n, Lang AE. Neuropathological Findings in choreaacanthocytosis: new insights into mechanisms behinding Parkinsonism and contractions. Acta Neuropatoli. (2014) 127:613–5. doi: 10.1007 / s00401-013-1241-3
PubMed Abstrakti / CrossRef kokoteksti / Google Scholar
15. Sorrentino G, De Renzo A, Miniello s, Nori O, Bonavita V. akantosyyttien myöhäinen ilmaantuminen koreaakantosytoosin aikana. J Neurol Sci. (1999) 163:175–8.
PubMed Abstract / Google Scholar
16. Bayreuther C, Borg M, Ferrero-Vacher C, Chaussenot A, Lebrun C. Choréo-acanthocytose sans acanthosytes. Revue Neurol. (2010) 166:100–3. doi: 10.1016 / J.neurol.2009.03.005
CrossRef Full Text | Google Scholar
17. Lesage S, Drouet V, Majounie E, Deramecourt V, Jacoupy M, Nicolas A, et al. Autosomaalisesti resessiivisessä parkinsonismissa vps13c-funktion häviäminen aiheuttaa mitokondrioiden toimintahäiriöitä ja lisää PINK1 / Parkin-riippuvaista mitofagiaa. Olen J. Hum Genet. (2016) 98:500–13. doi: 10.1016 / J.ajhg.2016.01.014
PubMed Abstract | CrossRef Full Text | Google Scholar
18. Gauthier J, Meijer IA, Lessel D, Mencacci NE, Krainc D, Hempel m, et al. Resessiiviset mutaatiot >vps13d aiheuttavat lapsuudessa alkavia liikehäiriöitä. Ann Neurol. (2018) 83: I6. doi: 10.1002 / ana.25204
PubMed Abstract | CrossRef Full Text | Google Scholar
19. Kurano Y, Nakamura M, Ichiba M, Matsuda M, Mizuno E, Kato m, et al. Koreiinin in vivo-jakelu ja lokalisointi. Biochem Biophys Res Commun. (2007) 353:431–5. doi: 10.1016 / j. bbrc.2006.12.059
PubMed Abstract | CrossRef Full Text | Google Scholar
20. Lang F, Pelzl L, Schöls L, Hermann A, Föller M, Schäffer TE, et al. Neuronit, erytrosyytit ja sen jälkeen-koreiinin monipuoliset toiminnot. Neurosignals (2017) 25:117-26. doi: 10.1159/000485457
PubMed Abstrakti / CrossRef kokoteksti / Google Scholar
21. Benninger F, Afawi Z, Korczyn AD, Oliver KL, Pendziwiat M, Nakamura m, et al. Kohtaukset esittävinä ja näkyvinä oireina chorea-acanthosytoosissa, jossa on C. 2343del VPS13A-geenimutaatio. Epilepsia (2016) 57:549-56. doi: 10.1111 / epi.13318
PubMed Abstract | CrossRef Full Text | Google Scholar