Frontiers in Pediatrics
- Introduction
- pitkän QT-ajan oireyhtymä
- Lqts1
- LQTS2
- LQTS3
- LQTS4
- LQTS5
- LQTS6
- LQTS7
- LQTS8
- LQTS9 ja LQTS10
- LQTS11
- LQTS12
- LQTS13
- hankittu LQTS
- Elektrokardiografiset piirteet pitkän QT-oireyhtymän kolmessa yleisessä muodossa
- genotyyppi-fenotyyppi
- LQTS: n kliininen hoito
- LQTS: Saudi Perspective
- Eturistiriitalausunto
- kiitokset
Introduction
Familiaaliset tai perinnölliset sydämen rytmihäiriöt sisältävät merkittäviä prosentteja rytmihäiriöistä ja myös syy-seuraus sydänperäisestä äkkikuolemasta (SCD) (1, 2). Viimeisten kahden vuosikymmenen aikana tutkijat ja kliinikot ovat tehneet valtavasti työtä selvittääkseen synnynnäisten familiaalisten rytmihäiriöiden (1-8) monimutkaisia ja monimutkaisia mekanismeja. Arytmogeneesin mekanismin ymmärtämiseksi meidän on tiedettävä sydämen solurakenteen perusteet ja niiden elektrofysiologiset ominaisuudet. Sydämen myosyytit ovat tärkeimmät toimivat solut sydämessä ja ne ovat laajasti kytketty niin, että impulssit etenevät nopeasti ja tasaisesti. Kardiomyosyytit erotetaan toisistaan erikoistuneella rajapinnalla, jota kutsutaan interkaloituneeksi levyksi; välilevyssä sijaitsevat aukkoliitosproteiinit, sydämen desmosomit ja ionikanavat (9). Gap liitokset koostuvat tiiviisti pakattu connexins jotka mahdollistavat solujen välisen vaihdon pieniä molekyylejä ja myös sallia virtaa herättäviä virtoja yhdestä solusta sen naapurisoluun. Desmosomit yhdessä adherens-liittymien kanssa ovat vastuussa yksittäisten kardiomyosyyttien mekaanisista liitteistä. Kaikki nämä osat intercalated levyt ovat peräkkäin eriytyneet ja jokainen komponentti suorittaa sen ainutlaatuinen tehtävä; häiriöt yhden komponentin vaikuttaa toimintaan muiden komponenttien, joka altistaa sydämen kehittää rytmihäiriöitä (9-11). Sydämen ionikanavat ovat huokosia muodostavia proteiinikomplekseja, jotka tarjoavat jännitteen aidattuja ja taidokkaasti koordinoituja ionivirtojen sisäänpäin ja ulospäin suuntautuvia liikkeitä solukalvojen poikki, mikä on välttämätöntä sydämen rytmin muodostumiselle ja etenemiselle. Pitkä QT-oireyhtymä (lqts), lyhyt QT-oireyhtymä (sqts), sairas sinus-oireyhtymä (SSS), sydämen johtumisvika (CCD), BrS, katekoliaminerginen polymorfinen kammiotakykardia (CPVT), varhainen repolarisaatio-oireyhtymä (ERS) ja familiaalinen eteisvärinä (Af) ovat tällä hetkellä tunnettuja sydämen kanavointipatioita, jotka voivat johtua yhdestä tai useammasta viasta sydämen rytmin muodostukseen ja lisääntymiseen liittyvissä geeneissä.
tässä katsauksessa kuvataan yksinomaan LQTS: ään liittyviä rytmihäiriöitä, sen patofysiologiaa ja tällä hetkellä saatavilla olevaa kliinistä hoitoa. Olemme olleet edelläkävijöitä geneettisen patologian selvittämisessä familiaalisissa sydämen rytmihäiriöissä Saudi-Arabiassa (1, 12-14), teemme edelleen kardiogeneettisiä tutkimuksia Saudi-Arabiassa, tämän tarkastelun lopussa keskustelemme tällä hetkellä saatavilla olevista tiedoista geneettisistä ja kliinisistä löydöksistä, jotka on saatu useista Saudi-Arabian perheistä, joilla on synkopeen ja SCD: n historia. Lisäämme myös äskettäin julkaistut tiedot LQTS: stä Riadissa sijaitsevan King Faisalin Erikoissairaalan ja tutkimuskeskuksen tiimiltä (15).
pitkän QT-ajan oireyhtymä
synnynnäinen LQTS on perinnöllinen sairaus, joka määritellään EKG: ssä (QT-ajan pitenemisenä). Potilaat, joilla on kaikki LQTS-muodot, ovat alttiita kammiotakyarytmialle, kääntyvien kärkien takykardialle (TDP), joka johtaa toistuvaan pyörtymiseen tai SCD: hen. Monissa tapauksissa pyörtyminen tai äkkikuolema voi olla ensimmäinen ja ainoa ilmentymä. LQTS vaikuttaa arviolta 1 2000 ihmistä maailmanlaajuisesti (16). LQTS: n tunnusmerkkinä on EKG: n QT-ajan piteneminen (korjattu sydämen syketiheydeksi eli QTc: ksi). QTc: n normaaliarvo on 440 ms miehillä ja 450 ms naisilla. Lapsilla ikä-ja sukupuolisidonnaisilla arvoilla on merkitystä. Viimeaikaiset konsensussuositus LQTS diagnoosi ovat seuraavat (17, 18):
1. LQTS on diagnosoitu:
a. jos LQTS-riskiarvo on ≥3, 5, jos QT-ajan pitenemiselle ei ole sekundaarista syytä ja/tai
B. Jos yhdessä lqts-geenissä on yksiselitteisesti patogeeninen mutaatio tai
c. Toistuvassa 12-johtoisessa elektrokardiogrammissa sydämen syketiheyden mukaan korjattu QT-aika (QTc) ≥500 ms ja QT-ajan pidentymiseen ei ole sekundaarista syytä.
2. LQTS voidaan diagnosoida QTc: n ollessa 480-499 ms toistuvassa 12-lyijyisessä ECGs: ssä potilaalla, jolla on selittämätön pyörtyminen ilman QT-ajan pidentymisen toissijaista syytä ja patogeenistä mutaatiota.
potilailla, joiden QTc-aika oli ≥500 ms, oli merkitsevästi suurempi kumulatiivinen todennäköisyys saada ensimmäinen pyörtyminen kuin potilailla, joiden QTc-aika oli <500 ms syntymästä 20 vuoden ikään (19). Mutta potilailla, joilla on esiintynyt 2, 3 ja 4 pyörtymisjaksoa, seuraavan pyörtymisjakson riski oli lähes sama potilailla, joilla oli kapea tai pitkittynyt QTc-aika (19). Lqts: n molekyyliperusta on heterogeeninen, ja tähän mennessä 13 eri geenissä tapahtuneiden mutaatioiden on kuvattu johtuvan lqts: stä (1-8, 19, 20). Geenivirhe löytyy yleensä 70% LQTS-potilaista jostakin näistä 13 geenistä (1-8, 19, 20). Niistä tällä hetkellä tiedossa 13 eri lqts, yleisimpiä ovat lqts1, LQTS2, ja lqts3, koska vikoja sydämen ionikanava geenit, kcnq1, KCNH2, ja SCN5A, vastaavasti. Yhdeksänkymmentä prosenttia kaikista lqts-kausaalisista mutaatioista löytyy näistä kolmesta geenistä (1-8, 19, 20). Jäljellä olevien 10 geenin mutaatiot ovat harvinaisia ja käsittävät vain ∼10% kaikista nykyisin tunnetuista LQTS-mutaatioista.
pitkä QT-oireyhtymä on yleensä autosomissa dominoiva sairaus, mutta joskus useita mutaatioita yhdessä geenissä tai eri geeneissä havaittiin 5-10%: lla LQTS-potilaista (21, 22). Potilailla, joilla on useita mutaatioita, voi esiintyä pidempi QTc kuin niillä, joilla on yksi mutaatio, ja näillä potilailla on myös 3, 5-kertainen riski henkeä uhkaaviin sydäntapahtumiin.(21, 22)
Lqts1
Kcnq1-geenin mutaatiot ovat kaikkien LQTS: ien yleisin muoto, ja niistä käytetään nimitystä lqts tyyppi 1 (lqts1). Viisikymmentä prosenttia kaikista lqts-kausaalisista mutaatioista löytyy kcnq1-geenistä (20). KvLQT1 (tunnetaan myös nimellä Kv7. 1) on kcnq1-geenin valmistama proteiini, joka muodostaa tetrameerejä solujen sisällä olevassa endoplasmaisessa retikulumissa, joka alustavasti kokoontuu proteiinin minkin kanssa (koodattuna KCNE1: llä) ja kuljetetaan sitten sydämen myosyyttien plasmakalvoon, jossa ne välittävät hitaasti aktivoivaa virtaa, joka kiihdyttää toimintakyvyn repolarisaatiota sydänkudoksissa, tätä virtaa kutsutaan IK: ksi (kuva 1). LQTS1 kausaaliset kcnq1-mutaatiot ovat useimmiten missense-mutaatioita ja harvoissa tapauksissa ne voivat olla kehyksensiirtomutaatioita C-terminaalisella alueella (23). Kcnq1: n mutaatiokantajien sydämen rytmihäiriöt laukaisevat adrenergiset asemat, esim. henkinen stressi, fyysinen rasitus, sukellus, uinti (24-26). Potilailla, joilla on mutaatioita KvLQT1: n transmembraanisissa domeeneissa, on suurempi LQTS: ään liittyvien sydäntapahtumien riski ja he ovat herkempiä sympaattiselle stimulaatiolle (26, 27). Potilailla, joilla on missense-mutaatioita, on suurempi riski kuin potilailla, joilla on ei-sense-tai typistäviä mutaatioita (27, 28). Myös kcnq1-geenin 3′ – UTR: n vaihtelu vaikuttaa rytmihäiriöalttiuteen merkittävästi, oletettavasti vaikuttamalla geenin ilmentymiseen (29). Potilaat, joilla on kcnq1-mutaatioiden aiheuttamia rytmihäiriöitä, reagoivat melko hyvin β-salpaajiin, mutta jotkut potilaat voivat silti olla vähemmän reagoivia tai jopa resistenttejä tälle lääkkeelle. Tuoreessa artikkelissa Barsheshet et al. (30) väitti, että potilaat, joilla on mutaatioita Sytoplasmasilmukan (C-loop) alueen ulkopuolella KvLQT1: ssä, reagoivat vähemmän β-salpaajiin.
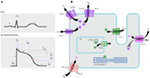
Kuva 1. Kammioperäiseen aktiopotentiaaliin vaikuttavat ioniset virrat (A) ja kardiomyosyytin kaavamainen esitys, jossa näkyvät (vain) ne proteiinit, jotka osallistuvat perittyjen rytmihäiriöoireyhtymien patogeneesiin (B). Kohdassa (A) vaikutuspotentiaali on linjassa sen likimääräisen vaikutusajan kanssa EKG: n aikana. Kohdassa (B) ei ole kuvattu ankyriini-B: tä, adapteriproteiinia, joka liittyy pitkän QT-oireyhtymän tyyppiin 4.
homotsygoottiset tai yhdisteen heterotsygoottiset mutaatiot kcnq1-geenissä, jotka voivat aiheuttaa taudin resessiivisen muodon, Jervellin ja Lange-Nielsenin oireyhtymän (jlns), tyypin 1 (31), ovat melko harvinaisia. JLNS-potilaat kärsivät myös vakavista sydämen rytmihäiriöistä ja kuuroudesta (31, 32). Potilailla, joilla on JLNS: ään johtuvia kcnq1-mutaatioita, ei yleensä ole mitään toiminnallista IK: ta (28, 31-33). On mahdollista, että joillakin potilailla ei ole kuuroutta huolimatta homotsygoottisista tai yhdisteen heterotsygoottisista mutaatioista kcnq1: ssä, tällaisia tapauksia kutsutaan autosomaalisesti resessiiviseksi lqts1: ksi (13, 32). Näillä potilailla voi edelleen esiintyä pieni määrä toiminnallista IK-virtaa (<10% IK: n kokonaismäärästä), joka ylläpitää kuulotoimintaa, mutta heidän sydämen rytmihäiriönsä ovat yhtä vakavia kuin JLNS-potilailla (13, 32).
LQTS2
tämän tyyppinen LQTS on yhtä yleinen kuin LQTS1, ja 35-40%: lla LQTS-potilaista, joilla oli havaittavissa oleva mutaatio (1, 20, 24, 25). KCNH2 koodaa HERG-proteiinia (Kv11. 1), joka on nopeasti aktivoivan viivästyneen tasasuuntaajan k+-virran (IKr) α-alayksikkö. Tämän kv11.1-kanavafunktiota vähentävän geenin patogeeniset mutaatiot pidentävät QT-ajan kestoa (Kuva 1) ja ovat syy-seuraus LQTS2: sta. 29 prosenttia lqts2: n pyörtymishyökkäyksistä tapahtui levon/unen aikana ja vain 13 prosenttia pyörtymishyökkäyksistä raportoitiin tapahtuvan harjoituksen aikana (25, 34). Äkilliset hätkähdyttävät äänet, kuten herätyskellojen, ovikellojen ja puhelinten soitot, laukaisevat näillä potilailla tyypillisesti pyörtymiskohtauksia (24, 34). Potilaat, joilla on mutaatioita kv11.1: n huokosmuodostusalueella (jota koodaa kcnh2-geeni), ovat alttiita suurelle rytmihäiriöihin liittyvien sydäntapahtumien riskille verrattuna potilaisiin, joilla on huokosettoman alueen mutaatioita (35). Vastasyntyneillä 2: 1 eteis-kammiokatkos (AV) liittyy ensisijaisesti KCNH2-mutaatioihin (36). LQTS: n komplisoimaa täydellistä AV-salpausta havaittiin myös 17%: lla aikuispotilaista, joilla oli kcnh2-geenin mutaatio (37). Homotsygoottiset mutaatiot kcnh2: ssa ovat harvinaisia ja kun niitä esiintyy, potilaat kärsivät vakavasta LQTS: n muodosta, jossa 2:1 AV-lohko ja vaikeat kammioperäiset rytmihäiriöt, kohdunsisäisten vaiheiden aikana sekä syntymän jälkeen (11, 38-40). Lqts2-patologiassa kuvattujen lukuisten mutaatioiden lisäksi myös kcnh2-geenin yleiset polymorfiset variantit saattoivat moduloida taudin vaikeusastetta. Kiehtova esimerkki on k897t polymorfismi (SNP) kcnh2: ssa, jota esiintyy 33%: lla väestöstä (41). K897T: n raportoitiin pahentavan KCNH2-mutaatioiden patogeenisuutta (42, 43).
LQTS3
Nav1.5 on jänniteriippuvaisen sydämen Na+-kanavan huokosia muodostava α-alayksikkö, se on scn5a-geenin koodaama integraalinen kalvoproteiini ja osallistuu sydämen toimintapotentiaalien käynnistämiseen ja johtamiseen. Sydämen Na+ – kanavat koostuvat huokosia muodostavasta α-alayksiköstä (koodattuna SCN5A: lla) ja yhdestä tai useammasta ylimääräisestä β-alayksiköstä.
LQTS3 johtuu α-alayksikön nopeaa inaktivaatiota häiritsevistä funktiomutaatioista (Kuva 1), ja SCN5A-geenin mutaatiota on kuvattu <10%: lla kaikista LQTS-potilaista, joilla on mutaatio (1, 20, 24). LQTS3-potilaat kokevat suurimman osan (39%) sydäntapahtumista unen/levon aikana (25) ja noin ∼13%: n tapauksista raportoitiin esiintyneen liikunnan aikana (25). Useissa tapauksissa yhden SCN5A-mutaation osoitettiin aiheuttavan kaksi tai jopa kolme eri rytmihäiriöiden fenotyyppiä samassa perheessä, esim.LQTS, BrS tai CCD (44-47). Miespotilaat, joilla on LQTS3-mutaatio, voivat saada oireita paljon aikaisemmin kuin naispotilaat (48). Sekä heterotsygoottisia että homotsygoottisia SCN5A-mutaatioita on kuvattu lqts3:ssa funktionaalisella 2: 1 AV-lohkolla (49). Olemme äskettäin löytäneet iranilaisen perheen, jolla on 1507_1509delQKP-mutaatio useissa perheenjäsenissä, joissa potilaat olivat yhdistäneet LQTS: n ja CCD: n (tietoja ei näy). Tätä mutaatiota on raportoitu myös muista maista tulevilla potilailla, mikä viittaa siihen, että kyseessä on toistuva ja hot spot-mutaatio . 1493delk – ja 1795insd-mutaatioissa (44, 51) raportoitiin myös sellaisia mutaatioita, joilla oli tällainen toimintakyvyn menetys-ja vahvistumisominaisuus aktiopotentiaalin eri vaiheissa.
joskus SNP: llä saattoi olla myös patogeeninen vaikutus kantajiinsa. S1103Y on scn5a-geenin yleinen muunnos, jota esiintyy 13 prosentilla afroamerikkalaisista (52). Tämän variantin kantajilla on lisääntynyt rytmihäiriöiden ja kätkytkuoleman (kätkytkuolema) riski (52).
LQTS4
LQTS4 edustaa LQTS: n ensimmäistä ei-kanavaista muotoa. Mutaatio ank2: ssa, adapteriproteiinissa, johtaa solunsisäiseen kalsiumin ylikuormitukseen, joka edistää LQTS4: ää (53, 54). QT-ajan pidentymisen lisäksi oireyhtymää sairastavilla potilailla voi olla sinusbradykardiaa, puuskittaista AF: ää ja CPVT: tä (54). ANK2-mutaatioiden patogeeninen vaikutus voi olla keskivaikea tai vaikea, ja kliiniset ilmaukset riippuvat mutaation vakavuudesta.
LQTS5
Kcne1: n mutaatiot liittyvät LQTS5: een (Kuva 1) (55, 56). Heterotsygoottiset KCNE1-mutaatiot pienentävät IKs: ää kohdistamalla dominoivan negatiivisen vaikutuksen siihen liittyvään normaaliin alleeliin ja johtavat viivästyneeseen sydämen repolarisaatioon (Kuva 1), mikä lisää rytmihäiriöiden riskiä (6). Potilaat, joilla on homotsygoottisia KCNE1-mutaatioita, kärsivät JLNS: stä (tyyppi 2) (57, 58).
D85N on polymorfismi kcne1-geenissä, joka esiintyy 0: ssa.7-1% väestöstä (41). Tutkimuksessa Nishio et al. (59), d85n-polymorfismia havaittiin useammin LQTS-potilailla, mikä tekee siitä riskigenotyypin LQTS-patologiassa (mahdollisesti vain Aasialaispopulaatiossa). Euroopassa D85N: ää raportoitiin 5%: lla lqts (alqts) – potilaista (32 potilaan kohortissa), joilla oli esiintynyt TdP: tä (60).
LQTS6
Kcne2-geeni koodaa minkkiin liittyvää peptidiä 1 (MiRP1), joka on sydämen kaliumkanavan ikr: n oletettu β-alayksikkö (Kuva 1). Mutaatiot KCNE2-geenissä voivat myös aiheuttaa vikoja viivästyneen tasasuuntaajan kaliumvirran (IKr) nopeasti aktivoivassa komponentissa, joka on lqts6: n (61) patologinen perusta. Kuulo / akustinen ärsyke, kuten herätyskellon melu, ovikello jne. voi aiheuttaa synkopaalihyökkäyksiä KCNE2-mutaation kantajissa, jotka ovat samanlaisia kuin KCNH2-mutaatio (62).
LQTS7
tämä oireyhtymä tunnetaan myös nimellä Andersen-Tawilin oireyhtymä (ATS). ATS on harvinainen sairaus, joka ilmenee satunnaisena pyörtymisenä ja sydänpysähdyksenä. EKG: n ominaisuuksiin kuuluvat lievä QT-ajan piteneminen, epänormaalit U-aallot, usein toistuva kammion ektopia, kaksisuuntainen kammiotakykardia (vt) ja polymorfinen VT. Tällä oireyhtymällä on myös ekstrasardiaalisia piirteitä, esim.luurankolihasten kausittainen halvaantuminen ja kehityshäiriöt, kuten suulakihalkio, matalat korvat, lyhytkasvuisuus ja raajojen kehityshäiriöt (63). Suurimmalla osalla kliinisesti diagnosoiduista ATS-potilaista raportoitiin kcnj2: n mutaatio (63). KCNJ2 koodaa sisäänpäin korjaavien kaliumkanavien huokosia muodostavan alayksikön (IK1) (Kuva 1) (64, 65).
LQTS8
, joka tunnetaan myös nimellä Timothyn oireyhtymä (TS), potilailla esiintyy voimakasta QT-ajan pitenemistä ECGs: ssä, johon liittyy syndaktyliaa, kaljuuntumista syntyessään ja pieniä hampaita 100%: ssa tapauksista sekä vähemmän läpäiseviä sydämen rakenteellisia epämuodostumia, kehitysvammaisuutta, autismia ja kasvojen dysmorfisia piirteitä (66). Alatyyppejä on kaksi: ts1 (klassinen) ja TS2 (harvinainen muoto).
TS2 on kardiologisesti ankarampi kuin TS1 (66, 67). TS2-potilailta puuttuu myös syndaktylia (67). Cacna1c-geeniä koodaavan L-tyypin kalsiumvirran (ICa-L) α-1-alayksikön mutaatiot johtavat TS: n molempiin muotoihin (LQTS8). Cacna1c-geenissä esiintyy vaihtoehtoisesti toisensa poissulkevia ekson-8: aa, selvyyden vuoksi ne nimetään ekson-8: ksi ja ekson-8A: ksi. sydämessä ja aivoissa, joissa cacna1c on pääasiassa ilmaistu, ekson-8 esiintyy ∼80%: ssa mRNA-transkripteistä ja ekson-8A esiintyy ∼20%: ssa transkripteistä (66). G406R: n mutaatio ekson-8A: ssa johtuu TS: n klassisesta muodosta (TS1) ja G402S: n ekson-8: ssa raportoitiin severerissä TS2: ssa. Uusi lisäys tähän luetteloon on Ala1473Gly-mutaatio, joka on kuvattu TS-lapsessa, jolla on edelleen laajentunut fenotyyppi (68). Kaikki mutaatiot olivat vanhemmassa joko de novo tai mosaiikki, ja kaikki ICa-L-kanavan toiminnan tulosvahvuus (66-69).
LQTS9 ja LQTS10
Cav3: n tai SCN4B: n mutaatiot saavat aikaan toimintakyvyn vahvistumisen myöhäisessä INa: ssa aiheuttaen lqts3: n kaltaisen fenotyypin (70-74). Ne tunnetaan nimillä LQTS9 (liittyy CAV3-mutaatioon) ja LQTS10 (liittyy SCN4B-mutaatioon).
luolat on kuvattu hienosti engelmanin et al. (72)” pieninä luolina ” plasmakalvossa. Ne ovat pieniä päällystämättömiä kuoppia, ja niitä pidetään tärkeiden dynaamisten ja säätelevien tapahtumien tapahtumapaikkana plasmakalvossa (72, 73). Kaveoliinit ovat luolien pääasiallisia proteiineja, ja caveolin-3: a (koodaa geeni CAV3) esiintyy erityisesti kardiomyosyyteissä ja luustolihassoluissa. Useiden sydämen ionikanavien on erityisesti raportoitu olevan lokalisoituja sydämen myosyyteistä erotetuissa luolissa, jotka ovat rikastuneet kaveolin-3: lla (72, 73). Lisäksi β-adrenergisen reseptorin signalointikaskadin komponentit ovat myös läsnä caveolae-rikasteisissa kalvoissa (72, 73).
SCN4B koodaa NaVß4: ää, joka on sydämen natriumkanavan β – alayksikkö. Tähän mennessä vain yksi mutaatio (L179F) on raportoitu tässä geenissä meksikolaisessa perheessä, jossa on useita sairastuneita perheenjäseniä (74). Tämän geenin mutaation havaittiin johtavan Nav1.5-virran (74) toiminnan vahvistumiseen.
LQTS11
sydämessä sydämen toimintapotentiaalin sympaattinen säätely (APD) välittyy β-adrenergisen reseptorin (β-AR) aktivaation kautta, mikä edellyttää AKAP9: n (Yotiao) yhdistämistä IKS-kanavan α-alayksikköön (KvLQT1). Mutaatio AKAP9: ssä aiheuttaa LQTS11 (75). Tähän mennessä on raportoitu vain yksi mutaatio, S1570L, AKAP9: ssä (75).
LQTS12
α-1-Syntrofiinin (SNTN1) geenin mutaatio johtuu LQTS12: sta. Tämän geenin mutaatio johtaa sydämen natriumkanavan (Nav1.5) toiminnan vahvistumiseen, joka on lqts12: n (76) patologinen perusta.
LQTS13
g proteiinikytkettyä, sisäisesti korjautuvaa kaliumkanavan alayksikköä (Kir3.4) koodaa KCNJ5-geeni. Funktion menetys mutaatio tässä geenissä voi aiheuttaa LQTS13 (77). Tähän mennessä on kuvattu vain yksi mutaatio, G387R, kiinalaisessa perheessä, jossa on yhdeksän potilasta, joilla on tämä mutaatio. KIR3.4: n alentunutta plasmakalvon ilmentymistä ehdotettiin LQTS: n patologiaksi potilailla.
hankittu LQTS
synnynnäisen LQTS: n lisäksi on olemassa myös toinen lqts: n muunnos, joka tunnetaan nimellä aLQTS, joka johtuu tekijöistä ja aineista, jotka vähentävät kaliumvirtaa ja heikentävät sydänlihaksen repolarisaatiokykyä. Hyvin tunnettuja tiloja ovat naissukupuoli, hypokalemia ja sydämen kaliumkanavia (78-80) estävät lääkkeet. Useat yleisesti määrätyt lääkkeet voivat myös ensisijaisesti sitoa ja estää HERG-kanavan (Kv11.1, kcnh2-geenin koodaama proteiini) ja altistaa aLQTS: lle (79, 80). Äskettäin osoitettiin, että IKs: n salpaus voi myös edistää lääkkeiden aiheuttamaa aLQTS: ää, erityisesti kun repolarisaatioreservi vaarantuu (81). Fluoksetiinin ja norfluoksetiinin havaittiin tukahduttavan IKs-ominaisuudet sekä in vivo että in vitro, ja ne johtivat merkittäviin LQTS-arvoihin (81). Polymorfismien, d85n minkeissä (geeni KCNE1), T8A, Q9E MiRP1: ssä (geeni KCNE2), jotka ovat ik-ja IKr-kanavien oletettuja β-alayksiköitä, on raportoitu aiheuttavan aLQTS: ää (82, 83). Autoimmuunisairaus LQTS: ää on raportoitu myös potilailla, joilla on IgG: tä, joka sisältää anti-HERG-vasta-aineita (84).
Elektrokardiografiset piirteet pitkän QT-oireyhtymän kolmessa yleisessä muodossa
tyypilliset ST-T-aaltomallit esiintyvät suurimmalla osalla potilaista, joilla on genotyyppi LQTS, ja niitä voidaan käyttää LQTS1 -, LQTS2-ja mahdollisesti lqts3-genotyyppien (85, 86) tunnistamiseen.
LQTS: n LQTS1-muoto liittyy laajaan T-aaltoon lyhentämättä QT-aikaa harjoituksessa (Kuva 2a). LQTS2 liittyy matalan amplitudin, usein bifid, t-aaltoihin (Kuva 2b). LQTS3 liittyy pitkään iso-sähköiseen segmenttiin ja kapeapohjaiseen, korkeaan T-aaltoon (Kuva 2C). Synnynnäisillä LQTS-potilailla ilmenevän TDP: n taukoriippuvuus on genotyyppispesifistä, sillä se on vallitseva LQTS2-ryhmässä, mutta puuttuu lähes kokonaan LQTS1-ryhmässä (87).

kuva 2. EKG-tallenteet potilailta, joilla on LQTS1, LQTS2 ja LQTS3. (A) 12-lyijyinen EKG 18-vuotiaalla miehellä, jolla on kcnq1-mutaatio. QT-aika pitenee (QTc = ±500 ms). ST-segmentillä on laaja pohja ja suhteellisen suuri amplitudi. Johtumisväli on normaali (standardikalibrointi). (B) 12-lyijyinen EKG 14-vuotiaalla tytöllä, jolla on kcnh2-mutaatio. QT-aika pitenee (QTc ± 520 ms). ST-segmentti on lovinen lyijy V3 ja on suhteellisen alhainen amplitudi raajajohtimet. Johtumisväli on normaali (standardikalibrointi). (C) 12-lyijyinen EKG 12-vuotiaalla pojalla, jolla on SCN5A-mutaatio. QT-aika pitenee (QTc ± 600 ms). ST-segmentissä on pitkä (lähes) iso-sähköinen segmentti, jossa on suuri, terävä ja kapea T-aalto. Johtumisväli on normaali (standardikalibrointi).
vaikka kuviot saattavat viitata LQTS: n tiettyyn genotyyppiin, poikkeuksia on havaittu usein.
genotyyppi-fenotyyppi
pitkä QT-oireyhtymä on autosomissa dominoiva sairaus. Heterotsygoottisten mutaatiokantajien genotyyppi-ja fenotyyppianalyysiä on tehty melko laajasti LQTS1 -, LQTS2-ja LQTS3-potilailla. Lapsuudessa sydäntapahtumien riski on merkitsevästi suurempi LQTS1-miehillä kuin LQTS1-naisilla, kun taas lqts2-ja LQTS3-potilailla (88-91) ei havaittu merkittäviä sukupuoleen liittyviä eroja sydäntapahtumien riskissä. Aikuisiällä (myös 40 ikävuoden jälkeen) LQTS1-ja LQTS2-naisilla saattaa olla merkitsevästi suurempi sydäntapahtumien riski kuin vastaavilla miehillä (88-91). Yleensä sydäntapahtumien kuolleisuus näyttää olevan vallitseva LQTS3-potilailla kuin LQTS1-ja LQTS2-potilailla (91). Naisilla, joilla on LQTS, on pienempi riski saada sydäntapahtumia raskauden aikana, mutta riski kasvaa huomattavasti 9 kuukauden synnytyksen jälkeen, erityisesti naisilla, joilla on mutaatio KCNH2-geenissä (92).
lasten sydänperäinen äkkikuolema voi johtua myös sydämen ionikanavageenien (93-96) mutaatioista. Noin 28% lapsista, joilla oli selittämätön SCD (1-18 vuotta, keski-ikä: 12,3 ± 3,8 vuotta) kantoi lqts-kausaalisten geenien mutaatioita (97). KÄTKYTKUOLEMISSA SCN5A: n mutaatiot näyttivät vallitsevilta (98, 99), mutta myös kcnq1: n, KCNH2: n, KCNE2: n ja CAV3: n, SCN4B: n ja SCN3B: n mutaatioita havaittiin (100, 101). Kohdunsisäisiä sikiökuolemia raportoitiin myös sydämen ionikanavageenien puutteiden vuoksi (12, 102).
LQTS: n kliininen hoito
kaikkien QT-aikaa pidentävien lääkkeiden lopettamisen ja myös elektrolyyttitasapainon korjaamisen ja/tai metabolisten tilojen sakkaamisen tulisi olla ensisijainen painopiste hoidettaessa (hankittuja) LQTS-potilaita. LQTS: n oireet ovat usein adrenergisiä, minkä vuoksi suositellaan yleensä rajoittamaan potilaiden osallistumista urheilutoimintaan (24, 25). LQTS: n kliinisen hoidon tukipilari on β-salpaus. Pitkävaikutteisia propranololi -, nadololi-ja metoprololivalmisteita käytetään yleensä, ja niiden tehoa β-Salpauksessa arvioidaan harjoituksen sykkeen bluntaamisella (esim.>20%) (24, 25). Kaikista β-salpaajista propranololia ja nadololia pidetään metoprololia parempina oireisilla potilailla (103). Lisäksi β-salpausta voidaan käyttää myös profylaktisena hoitona äänettömillä mutaatiokantajilla SCD: n (24) vähentämiseksi. Tutkimuksessa Barsheshet et al. (30) potilailla, joilla oli C-loop missense-mutaatioita KCNQ1-geenissä, oli suuri riski hengenvaarallisiin sydäntapahtumiin ja he saivat merkittävää hyötyä β-salpaajahoidosta. Koska LQTS2-potilailla on suurentunut riski 9 kuukauden synnytyksen jälkeen, beetasalpaajia tulee määrätä vähentämään sydäntapahtumia tämän korkean riskin jakson aikana (92). Implantoitavaa kardioverter-defibrillaattoria (ICDs) voidaan harkita potilaille, joilla on toistuva pyörtyminen β-salpaajahoidosta huolimatta, tai potilaille, joilla on suuri sydänpysähdysriski (esim., oireinen LQTS2 ja LQTS3, joihin liittyy dokumentoitu QTc-ajan piteneminen). Vasemman sydämen sympaattista denervaatiota (lcsd) suositellaan korkean riskin LQTS-potilaille, joilla ICD on vasta-aiheinen tai kieltäytynyt ja β-salpaajat eivät joko ole tehokkaita, eivät siedä, eivät hyväksy tai vasta-aiheisia (18, 104). JLNS-potilailla QTc on yleensä >500 ms ja heillä on myös suuri riski, β-salpaajien teho ei ole riittävä näillä potilailla, joille suositeltiin varhaisen vaiheen hoitoa ICD: hen (18, 31).
LQTS: Saudi Perspective
ensimmäinen raportti Saudi-Arabian LQTS: stä julkaistiin vuonna 1993 Riadin asevoimien sairaalasta (105). Yhdestä perheestä neljällä 6-48 kuukauden ikäisellä pikkulapsella ja pikkulapsella, joilla oli ollut toistuvia kohtauksia, todettiin LQTS (105). Sukuhistoria paljasti, että kahdella muulla laajennetulla perheenjäsenellä oli samanlaisia äkillisiä tajunnan menetyksiä ja kolme perheenjäsentä kuoli äkillisesti (105). Kaikissa tapauksissa alkudiagnoosi oli epilepsia (105). Useita vuosia myöhemmin on raportoitu kahdesta satunnaisesta tapausraportista, joissa vastasyntyneiden LQTS:n muunnos on verrattain vakavampi ja 2: 1 AV: n esto yhdistetty (106, 107). Kaikissa näissä julkaistuissa kliinisissä raporteissa ei ollut geneettisiä löydöksiä, jotka voisivat selittää LQTS: n patofysiologiaa niissä (105-107).
olemme ensimmäistä kertaa raportoineet geenivirheistä LQTS: n patologisena perustana vastaavilla potilailla Saudi-Arabiasta. Olemme tutkineet kuutta Saudiperhettä, joilla on ollut pyörtymisiä ja äkillisiä selittämättömiä sikiön, vastasyntyneen ja lasten (12-14) kuolemia. Autosomaalinen resessiivinen LQTS1 diagnosoitiin kahden perheen lapsilla (kuva 3). Autosomaalinen resessiivinen LQTS2 diagnosoitiin kahdessa perheessä (Kuvio 4). Eräässä perheessä, naispotilaalla diagnosoitiin autosomaalinen dominantti LQTS2 (kuva 5), potilaalla oli synkopaalikohtauksia synnytyksen jälkeisen toipumisjakson aikana sairaalassa, mikä on hyvin yleistä kcnh2-mutaation kantajilla naisilla. Kaikissa potilaissamme patogeenisten mutaatioiden tunnistaminen lqts-kausaalisissa sydämen ionikanavageeneissä johti vahvistettuun kliiniseen diagnoosiin, joka diagnosoitiin väärin epileptisiksi kouristuksiksi ennen kuin se lähetettiin meille (13, 14) äskettäin, Shinwari et al. (15) King Faisal Specialist Hospital and Research Centre raportoi lqts1 syy kcnq1 mutaatio, H258P, suuressa perheessä 12 vaikuttaa yksilöiden. Vain kaksi kantajaa oireili, niiden QTc oli >500 ms, ja β-salpaajat tukahduttivat kliiniset oireet yhdellä potilaalla ja toinen oireinen potilas tarvitsi ICD: n.
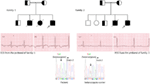
kuva 3. Sukutaulu perheen-1 ja 2 autosomaalinen resessiivinen LQTS1, probands näytetään nuolella. ECGs alkaen probands kahdessa perheessä on esitetty keskellä, jossa QTc 557 ja 529 ms, vastaavasti. Introninen mutaatio, c. 387-5 T > a KCNQ1-geenissä havaittiin potilailla (alla). Mutaatio näytettiin nuolella. Täyttämättömät ympyrät ja neliöt eivät kanna mutaatiota. Sairastuneet henkilöt näkyvät täytettyinä ympyröinä (nainen) ja neliöinä (mies). Puolitäytteiset neliöt ja ympyrät ovat yksilöitä, joilla on heterotsygoottimutaatio. Kuolleet yksilöt merkitään viilloilla, probandit merkitään nuolella ja konsanguineous avioliitto merkitään = Ekson − intron raja näkyy pisteviivalla, jossa nuoli osoittaa kohti eksonia.
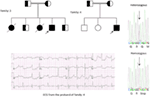
Kuva 4. Sukutaulu piirustus perheen-3 ja 4 autosomaalinen resessiivinen LQTS2. Perhe-4: n probandin EKG on esitetty sukutaulun alla, jossa näkyy sinustakykardia, lähes täydellinen AV-lohko, laaja monimutkainen pakorytmi hyvin pitkillä QTc-väleillä (QT >600 ms). Mutaatio, C. 3208 C > T (P. Q1070X) kcnh2-geenissä havaittiin potilailla (kuvassa oikealla) nuolella merkittynä.
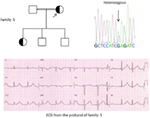
kuva 5. Ylhäällä vasemmalla: sukutaulu-5. Kyseinen henkilö näkyy täytetyn ympyrän (naaras) Proband on merkitty nuolella. 12-Lyijy-EKG proband (I:2). EKG näyttää korkean huipun, laajapohjaisen, suuren amplitudin T-aallon, QTc-aika on 580 ms. Kcnh2-geenin seulonta osoittaa nukleotidin “G” substituution “A”: lle (c.2362g > a, nuoli merkitty), mikä johtaa aminohapposubstituutioon, s.E788K.
geneettiset ja kliiniset löydökset tutkimuksessamme (12-14) Saudi-Arabiasta ovat varsin kiehtovia useista syistä: (1) yhteensä olemme tutkineet kuusiperheitä, joista neljä oli homotsygootteja/heterotsygootteja mutaatioille ja mutaatiot olivat peräisin esi-isien lähteestä.; (2) kaikki lqts: n mutaatiot olivat uusia, raportoitiin vain näissä Arabiperheissä; (3) mutaatioiden homotsygositeetin tai yhdisteen heterotsygositeetin vuoksi kliiniset fenotyypit olivat myös vakavia tutkituissa perheissämme (12-14). Olemme esittäneet, että geneettiset ja fenotyyppiset havainnot johtuvat Saudi-Arabiassa solmittujen verisukulaisavioliittojen äärimmäisen suuresta määrästä (108, 109). Tutkimuksemme tarjosi ensimmäisen tieteellisen näytön verisukulaisuuden roolista keskeisessä roolissa Selittämättömissä sydämen rytmihäiriöissä ja SCDs: ssä lapsilla ja nuorilla Saudi-Arabiassa (12-14). Olemme myös osoittaneet, että Kcnq1: n mutaatio c.387 -5 t > a (Nm_000218) (kuva 3) oli levinnyt Assirin maakunnassa Saudi-Arabiassa yhteisestä esi-isästä useiden sukupolvien aikana johtuen verisukulaisavioliittojen suuresta esiintymistiheydestä . Tähän mennessä kyseessä on Saudi-Arabian väestön yleisin lqts1-Kausaalinen mutaatio, joka havaittiin myös Kuningas Faisalin erikoissairaalassa ja Khamis Mashaytin sotilassairaalassa (julkaisematon). Samanlainen tulos saatiin myös lqts2-kausaalimutaatiolle, c. 3208 C > T (p.Q1070X) (Kuva 4) Kcnh2-geenissä (12, 14), joka on myös Saudi-Arabian perustajamutaatio ja eriytynyt useiden sukupolvien aikana Assirin alueella (katso kartta, kuva 6). Ennustamme, että on olemassa huomattava määrä yksilöitä, joilla on mainittu esi/perustaja mutaatiot (sekä kcnq1 ja KCNH2 ja muut geenit) tällä alueella ja myös suurissa kaupungeissa, kuten Riyadh, Jeddah ja Dammam johtuen kaupunkien muuttoliike. Enemmän perustaja tai esivanhempien mutaatioita patogeenisiä Lqts ovat hyvin paljon kaavaillaan muissa maakunnissa Saudi-Arabian johtuen korkea määrä verisukulaisia avioliittoja.
kuva 6. Saudi-Arabian kartta (Wikipedia). Assirin alue on merkitty paksulla viivalla, josta on löydetty perustajamutaatiot kcnq1-ja kcnh2-geeneissä, jotka on kuvattu suvuissa 1-4.
koska LQTS on autosomaalinen dominoiva sairaus, odotimme, että potilailla olisi pääasiassa heterotsygoottisia mutaation kantajia. Mutta tutkimuksessamme tunnistimme pääasiassa potilaita, joilla oli resessiivinen mutaatio (12-14). On selvää, että nämä potilaat tulevat ensin oireilemaan ja ohjattiin pääasiassa prinssi Sultanin sydänkeskukseen, jolloin he ovat meidän huomiomme kohteena. Kuitenkin havainnot tutkimuksistamme viittaa siihen, että resessiivinen LQTS voi olla kuolemaan kliinisiä fenotyyppejä lapsilla, ja ne eivät ole harvinaisia Saudi-Arabiassa (12-14). Koska heterotsygoottiset mutaationkantajat ovat myös alttiita rytmihäiriöille ja sen komplikaatioille, on tehtävä yhteinen aloite, jotta paikalliset Yleislääkärit, kardiologit ja kliiniset geneetikot saadaan yhteiselle foorumille tunnistamaan riskiryhmään kuuluvat henkilöt. Lisäksi tällä hetkellä meillä ei ole myöskään geneettistä tietoa muista familiaalisista rytmihäiriöistä, kuten CPVT: stä, SQTS: stä, BrS: stä, AF: stä jne. Erikoistuneet kardiogeneettiset keskukset pitäisi tehdä aloite etsiä geneettisiä vikoja, mutaatioita, ja suorittaa genotyyppi-fenotyyppi tutkimuksia kaikissa muodoissa perinnöllisiä rytmihäiriöitä. On myös otettava huomioon, että kaikilla geneettisillä rytmihäiriöillä ei ole sukuhistoriaa, koska monissa tapauksissa rytmihäiriöiden syy-seuraus-mutaatio on de novo-alkuperää, eli proband on ensimmäinen potilas kyseisessä perheessä, jolla on mutaatio, ja hän/hän on lähde, joka siirtää mutaation myöhempiin sukupolviin (110, 111). Koska Saudi-Arabiassa solmitaan hyvin paljon verisukulaisavioliittoja, odotamme paljon enemmän perustajamutaatioita, joilla on ratkaiseva rooli synnynnäisissä rytmihäiriöissä tässä maassa. Näiden perustajamutaatioiden tunnistaminen pitäisi olla meidän ensisijainen tehtävämme, joka helpottaisi meitä kehittämään tehokasta esiaviollista ja myös esioireista geneettistä neuvontaa tässä maassa. Mutaatiot lqts syy geenit voivat antaa vaihtelua kliinisen penetrance, yhdessä ääripäässä, jotkut kantajat voivat olla oletettavasti täysin terveitä, mutta jotkut kantajat voivat olla niiden ensimmäinen ilmentymä taudin pyörtyminen tai äkkikuolema. Nuoren lapsen tai aikuisen äkillinen kuolema aiheuttaa paljon psykologista ja emotionaalista taakkaa perheelle, kantajien seulonta on välttämätöntä, koska on olemassa yksinkertainen lääkitys, esim.β-salpaajat (myös käyttäytymisen muuttaminen), joka voisi hyvin tehokkaasti estää kantajia rytmihäiriöiden ja SCDs: n kohtalokkaista seurauksista. Henkilöillä, joilla on homotsygoottisia mutaatioita lqts-kausaalisissa geeneissä, voi olla vakava sairastuvuus ja äärimmäisen korkea kuolleisuus, mukaan lukien sikiön brady-takyarytmiat, ja monissa tapauksissa keskenmenoja, keskenmenoja raskaana olevilla äideillä (12-14).
Saudi-Arabiassakaan ei ole juurikaan tutkittu erilaisten SNP: iden esiintyvyyttä rytmihäiriöihin liittyvissä geeneissä ja myös niitä säätelevissä geeneissä. Splawski ym. (112) kuvattu yleinen s1103y-muunnos SCN5A-geenissä, joka liittyy rytmihäiriöihin afroamerikkalaisilla. Muunnos-alleeli, jota kutsutaan Y1103: ksi, kiihdyttää kanavan aktivoitumista, mikä lisää sydämen rytmihäiriöiden todennäköisyyttä afrikkalaista syntyperää olevilla henkilöillä (52, 112). K393N on variantti KCNQ1 geeni raportoitu LQTS1 potilailla Yhdysvalloissa, mutta, Arabipopulaatiossa olemme havainneet tämän variantin 2% yksilöistä (julkaisemattomia tietoja). Se, onko K393n-muunnos Kcnq1-geenissä arabeilla verrattavissa S1103y-muunnokseen (SCN5A) afroamerikkalaisilla tai D85n-muunnokseen (Kcne1) japanilaisella väestöllä, voisi myös olla tutkimuksen arvoinen (59).
Eturistiriitalausunto
kirjoittajat toteavat, että tutkimus tehtiin ilman kaupallisia tai taloudellisia suhteita, joita voitaisiin pitää mahdollisena eturistiriitana.
kiitokset
vilpittömät kiitoksemme Prof.Arthur Wildelle, Prof. Connie Bezzinalle ja “Heart” – lehden julkaisijalle heidän luvallaan käyttää tämän käsikirjoituksen lukua 1 viite. (113).
1. Bhuiyan ZA. Perinnöllisten Rytmihäiriöoireyhtymien kliininen ja geneettinen kirjo. Amsterdam: Amsterdamin Yliopisto (2009).
2. Priori SG, Aliot E, Blømstrom-Lundqvist C, Bossaert L, Breithardt G, Brugada P, et al. Task force on sudden Heart death, European society of cardiology. Europace (2002) 4:3-18. doi: 10.1053 / eupc.2001, 0214
CrossRef Koko Teksti
3. Wang Q, Shen J, Splawski I, Atkinson D, Li Z, Robinson JL, et al. SCN5A-mutaatiot, jotka liittyvät periytyvään sydämen rytmihäiriöön, pitkän QT-ajan oireyhtymään. Cell (1995) 80: 805-11. doi:10.1016/0092-8674(95)90359-3
PubMed Abstrakti / Pubmed kokoteksti | CrossRef kokoteksti
4. Curran minä, Splawski I, Timothy KW, Vincent GM, Green ED, Keating MT. Sydämen rytmihäiriöiden molekyyli: HERG-mutaatiot aiheuttavat pitkän QT-oireyhtymän. Cell (1995) 80: 795-803. doi: 10.1016/0092-8674(95)90358-5
PubMed Abstrakti / Pubmed Kokoteksti | CrossRef Kokoteksti
5. Wang Q, Curran ME, Splawski I, Burn TC, Millholland JM, VanRaay TJ, et al. Uuden kaliumkanavageenin positionaalinen Kloonaus: KVLQT1-mutaatiot aiheuttavat sydämen rytmihäiriöitä. Nat Genet (1996) 12:17-23. doi: 10.1038/ng0196-17
Pubmed Abstrakti / Pubmed koko teksti | CrossRef koko teksti
6. Splawski I, Tristani-Firouzi M, Lehmann MH, Sanguinetti MC, Keating MT. Hmink-geenin mutaatiot aiheuttavat pitkän QT-oireyhtymän ja estävät IKs: n toiminnan. Nat Genet (1997) 17:338-40. doi: 10.1038 / ng1197-338
Pubmed Abstrakti / Pubmed koko teksti | CrossRef koko teksti
7. Sanguinetti MC, Jiang C, Curran ME, Keating MT. Mekanistinen yhteys perityn ja hankitun sydämen rytmihäiriön välillä: HERG koodaa ikr-kaliumkanavan. Cell (1995) 81: 299-307. doi:10.1016/0092-8674(95)90340-2
PubMed Abstrakti / Pubmed kokoteksti | CrossRef kokoteksti
8. Neyroud N, Tesson F, Denjoy I, Leibovici M, Donger C, Barhanin J, et al. Uusi mutaatio kaliumkanavageenissä KVLQT1 aiheuttaa Jervellin ja Lange-Nielsenin kardioauditorisen oireyhtymän. Nat Genet (1997) 15:186-9. doi: 10.1038/ng0297-186
Pubmed Abstrakti / Pubmed koko teksti | CrossRef koko teksti
9. Kucera JP, Rohr s, Rudy Y. natriumkanavien lokalisointi interkalatoituihin levyihin moduloi sydämen johtumista. Circ Res (2002) 91:1176-82. doi: 10.1161 / 01.RES.0000046237.54156. 0 a
Pubmed Abstrakti / Pubmed koko teksti / CrossRef koko teksti
10. Oxford EM, Musa H, Maass K, Coombs W, Taffet SM, Delmar M. Connexin43 remodeling caused by inhibition of plakophilin-2 expression in heart cells. Circ Res (2007) 101:703-11. doi: 10.1161 / CIRCRESAHA.107. 154252
Pubmed Abstrakti / Pubmed Koko Teksti | CrossRef Koko Teksti
11. Rohr S. Molekylaarinen ylikuuluminen mekaanisten ja sähköisten liittymien välillä interkaloidun levyn kohdalla. Circ Res (2007) 101:637-9. doi: 10.1161 / CIRCRESAHA.107. 161901
CrossRef Koko Teksti
12. Bhuiyan ZA, Momenah TS, Gong Q, Amin AS, Ghamdi SA, Carvalho JS, et al. Toistuva sikiön kohdunsisäinen menetys, joka johtuu herg: n puuttumisesta: homotsygoottisen nonsense herg Q1070X-mutaation kliininen ja toiminnallinen Luonnehdinta. Heart Rhythm (2008) 5:553-61. doi: 10.1016 / J. hrthm.2008. 01. 020
Pubmed Abstrakti | Pubmed Koko Teksti | CrossRef Koko Teksti
13. Bhuiyan ZA, Momenah TS, Amin TS, Al-Khadra AS, Alders M, Wilde AAM, et al. Introninen mutaatio, joka johtaa ekson-2: n puutteelliseen ohitukseen kcnq1: ssä, pelastaa kuulemisen Jervellin ja Lange-Nielsenin oireyhtymässä. Prog Biophys Mol Biol (2008) 98:319-27. doi: 10.1016 / j.pbiomolbio.2008. 10. 004
Pubmed Abstrakti | Pubmed Koko Teksti | CrossRef Koko Teksti
14. Bhuiyan ZA, Al-Shahrani S, Al-Khadra AS, Al-Ghamdi s, Al-Kalaf K, Mannens MMAM, et al. Saudi-Arabian kuuden perheen lasten pitkän QT-oireyhtymän kliininen ja geneettinen analyysi: ovatko he erilaisia? Pediatr Cardiol (2009) 30:490-501. doi: 10.1007 / s00246-008-9377-y
Pubmed Abstrakti / Pubmed koko teksti | CrossRef koko teksti
15. Shinwari ZM, Al-Hazzani A, Dzimiri N, Tulbah S, Mallawi Y, Al-Fayyadh m, et al. Uuden kcnq1-mutaation tunnistaminen suuressa Saudiperheessä, jolla on pitkä QT-oireyhtymä: clinical consequences and preventive implications. Clin Genet (2013) 83: 370-4. doi: 10.1111 / j.1399-0004.2012.01914.x
Pubmed Abstrakti / Pubmed koko teksti | CrossRef koko teksti
16. Schwartz PJ, Stramba-Badiale M, Crotti L, Pedrazzini M, Besana a, Bosi G, et al. Synnynnäisen pitkä-QT-oireyhtymän esiintyvyys. Levikki (2009) 120:1761-7. doi: 10.1161 / CIRCULATIONAHA.109.863209
Pubmed Abstrakti / Pubmed Koko Teksti | CrossRef Koko Teksti
17. Schwartz PJ, Moss AJ, Vincent GM, Crampton RS. Pitkän QT-syndrooman diagnostiset kriteerit. Päivitys. Levikki (1993) 88:782-4. doi: 10.1161 / 01.CIR.88. 2. 782
CrossRef Koko Teksti
18. Priori SG, Wilde AA, Horie M, Cho Y, Behr ER, Berul C, et al. Tiivistelmä: HRS / EHRA / APHRS consensus statement on the diagnosis and management of patients with peritty primary arythmy syndromes. Europace (2013) 15:1389-406. doi: 10.1093 / europace/eut272
CrossRef koko teksti
19. Liu JF, Jons C, Moss AJ, McNitt S, Peterson DR, Qi m, et al. Syndroomarekisteri. Toistuvan pyörtymisen ja sitä seuranneiden kuolemaan tai lähes kuolemaan johtaneiden tapahtumien riskitekijät lapsilla ja nuorilla, joilla on pitkä QT-oireyhtymä. J Am Coll Cardiol (2011) 57:941-50. doi: 10.1016 / J.jacc.2010. 10. 025
Pubmed Abstrakti / Pubmed Kokoteksti | CrossRef Kokoteksti
20. Splawski I, Shen J, Timothy KW, Lehmann MH, Priori S, Robinson JL, et al. Mutaatioiden kirjo pitkän QT-oireyhtymän geeneissä. KVLQT1, HERG, SCN5A, KCNE1 ja KCNE2. Levikki (2000) 102:1178-85. doi: 10.1161 / 01.CIR.102. 10. 1178
Pubmed Abstrakti | Pubmed Koko Teksti | CrossRef Koko Teksti
21. Westenskow P, Splawski I, Timothy KW, Keating MT, Sanguinetti MC. Yhdistelmämutaatiot: yleinen syy vaikeaan pitkän QT-ajan oireyhtymään. Levikki (2004) 109:1834-41. doi: 10.1161 / 01.CIR.0000125524. 34234. 13
Pubmed Abstrakti / Pubmed Koko Teksti | CrossRef Koko Teksti
22. Mullally J, Goldenberg I, Moss AJ, Lopes CM, Ackerman MJ, Zareba W, et al. Hengenvaarallisten sydäntapahtumien riski potilailla, joilla on pitkä QT-oireyhtymä ja useita mutaatioita. Sydämen Rytmi (2013) 10:378-82. doi: 10.1016 / J. hrthm.2012. 11. 006
Pubmed Abstrakti | Pubmed Kokoteksti | CrossRef Kokoteksti
23. Napolitano C, Priori SG, Schwartz PJ, Bloise R, Ronchetti E, Nastoli J, et al. Genetic testing in the long QT syndrome: development and validation of a efficient approach to genotyping in clinical practice. JAMA (2005) 294:2975-80. doi: 10.1001 / jama.294.23.2975
Pubmed Abstrakti / Pubmed Koko Teksti | CrossRef Koko Teksti
24. Roden DM. Harjoittelu. Pitkän QT-ajan oireyhtymä. N Engl J Med (2008) 358: 169-76. doi: 10.1056/NEJMcp0706513
CrossRef koko teksti
25. Schwartz PJ, Priori SG, Spazzolini C, Moss AJ, Vincent GM, Napolitano C, et al. Genotyyppi-fenotyyppikorrelaatio long-QT-oireyhtymässä: geenispesifiset laukaisijat hengenvaarallisille rytmihäiriöille. Levikki (2001) 103:89-95. doi: 10.1161 / 01.CIR.103. 1. 89
CrossRef Koko Teksti
26. Ackerman MJ, testaaja DJ, Porter CJ. Uinti on geenispesifinen rytmihäiriö, joka laukaisee periytyvän pitkän QT-oireyhtymän. Mayo Clin Proc (1999) 74: 1088-94. doi:10.4065/74.11.1088
PubMed Abstrakti / Pubmed kokoteksti | CrossRef kokoteksti
27. Kapa S, Tester DJ, Salisbury BA, Harris-Kerr C, Pungliya MS, Alders M, et al. Genetic testing for long-QT syndrome: distincting pathogenic mutations from benign variants. Levikki (2009) 120:1752-60. doi: 10.1161 / CIRCULATIONAHA.109.863076
Pubmed Abstrakti / Pubmed Koko Teksti | CrossRef Koko Teksti
28. Moss AJ, Shimizu W, Wilde AA, Towbin JA, Zareba W, Robinson JL, et al. Tyypin 1 pitkän QT-syndrooman kliiniset näkökohdat kcnq1-geenin mutaatioiden sijainnin, koodaustyypin ja biofysikaalisen toiminnan mukaan. Levikki (2007) 115:2481-9. doi: 10.1161 / CIRCULATIONAHA.106. 665406
Pubmed Abstrakti / Pubmed Koko Teksti | CrossRef Koko Teksti
29. Amin AS, Giudicessi JR, Tijsen AJ, Spanjaart AM, Reckman YJ, Klemens CA, et al. Muunnokset kcnq1-koodatun Kv7.1-kaliumkanavan 3 ‘ – kääntämättömällä alueella muuttavat taudin vaikeusastetta potilailla, joilla on tyypin 1 pitkä QT-oireyhtymä, alleelispesifisesti. EUR Heart J (2012) 33:714-23. doi: 10.1093 / eurheartj / ehr473
Pubmed Abstrakti / Pubmed kokoteksti | CrossRef kokoteksti
30. Barsheshet A, Goldenberg I, O-Uchi J, Moss AJ, Jons C, Shimizu w, et al. Kcnq1-kanavan sytoplasmasilmukoiden mutaatiot ja hengenvaarallisten tapahtumien riski: vaikutukset β-salpaajahoidon mutaatiospesifiseen vasteeseen tyypin 1 pitkän QT-oireyhtymän hoidossa. Levikki (2012) 125:1988-96. doi: 10.1161 / CIRCULATIONAHA.111. 048041
Pubmed Abstrakti / Pubmed Koko Teksti | CrossRef Koko Teksti
31. Schwartz PJ, Spazzolini C, Crotti L, Bathen J, Amlie JP, Timothy K, et al. The Jervell and Lange-Nielsen syndrome: natural history, molecular basis, and clinical outcome. Levikki (2006) 113:783-90. doi: 10.1161 / CIRCULATIONAHA.105.592899
Pubmed Abstrakti / Pubmed Koko Teksti | CrossRef Koko Teksti
32. Bhuiyan ZA, Wilde AA. IKs sydämessä ja kuulo, korva voi tehdä vähemmän kuin sydän. Circ Cardiovasc Genet (2013) 6:141-3. doi: 10.1161 / SIRKGENETIIKKA.113. 000143
CrossRef Koko Teksti
33. Wollnik B, Schroeder BC, Kubisch C, Esperer HD, Wieacker P, Jentsch TJ. Periytyvissä sydämen rytmihäiriöissä esiintyvien dominoivien ja resessiivisten KVLQT1 K+ – kanavamutaatioiden patofysiologiset mekanismit. Hum Mol Genet (1997) 6:1943-9. doi: 10.1093/hmg / 6.11.1943
CrossRef Koko Teksti
34. Wilde AA, Jongbloed RJ, Doevendans PA, Düren DR, Hauer RN, van Langen IM, et al. Kuuloärsykkeet rytmihäiriöiden laukaisevina tekijöinä erottavat HERG-potilaat (LQTS2) KVLQT1-potilaista (LQTS1). J Am Coll Cardiol (1999) 33:327-32. doi: 10.1016 / S0735-1097(98)00578-6
yliviivataan koko teksti
35. Moss AJ, Zareba W, Kaufman ES, Gartman E, Peterson DR, Benhorin J, et al. Lisääntynyt rytmihäiriötapahtumien riski pitkän QT-ajan oireyhtymässä, johon liittyy mutaatioita ihmisen eetteri-a-go-go-sukuisen geenin kaliumkanavan huokosalueella. Levikki (2002) 105:794-9. doi: 10.1161 / hc0702.105124
Pubmed Abstrakti / Pubmed koko teksti | CrossRef koko teksti
36. Lupoglazoff JM, Denjoy I, Villain E, Fressart V, Simon F, Bozio A, et al. Vastasyntyneen pitkä QT-oireyhtymä: herg-mutaatioihin liittyvät johtumishäiriöt ja kcnq1-mutaatioihin liittyvä sinusbradykardia. J Am Coll Cardiol (2004) 43:826-30. doi: 10.1016 / J.jacc.2003.09.049
Pubmed Abstrakti / Pubmed Koko Teksti | CrossRef Koko Teksti
37. Chevalier P, Bellocq C, Millat G, Piqueras E, Potet F, Schott JJ, et al. Torsades de pointes complicating atrioventricular block: evidence for a genetic alttius. Sydämen Rytmi (2007) 4: 170-4. doi: 10.1016 / J. hrthm.2006. 10. 004
Pubmed Abstrakti | Pubmed Koko Teksti | CrossRef Koko Teksti
38. Hoorntje T, Alders M, van Tintelen P, van der Lip K, Sreeram N, van der Wal A, et al. Homotsygoottinen ennenaikainen herg-proteiinin katkaisu: ihmisen herg-tyrmäys. Levikki (1999) 100:1264-7. doi: 10.1161 / 01.CIR.100. 12. 1264
Pubmed Abstrakti | Pubmed Koko Teksti | CrossRef Koko Teksti
39. Piippo K, Laitinen P,Joutsen H, Toivonen L, Viitasalo M, Pasternack M, ym. Homotsygoottisuus herg-kaliumkanavamutaatiolle aiheuttaa vakavan pitkän QT-oireyhtymän muodon: selvän perustajamutaation tunnistaminen suomalaisilla. J Am Coll Cardiol (2000) 35: 1919-25. doi: 10.1016 / S0735-1097(00)00636-7
PubMed Abstrakti / Pubmed kokoteksti | CrossRef kokoteksti
40. Johnson Wh Jr, Yang P, Yang T, Lau YR, Mostella BA, Wolff DJ, et al. Kliininen, geneettinen ja biofyysinen karakterisointi homotsygoottisesta HERG-mutaatiosta, joka aiheuttaa vakavan vastasyntyneen pitkän QT-syndrooman. Pediatr Res (2003) 53:744-8. doi: 10.1203 / 01.PDR.0000059750.17002.B6
Pubmed Abstrakti / Pubmed Koko Teksti / CrossRef Koko Teksti
41. Ackerman MJ, testaaja DJ, Jones GS, will ML, Burrow CR, Curran ME. Etniset erot sydämen kaliumkanavavarianteissa: vaikutukset geneettiseen alttiuteen sydänperäiselle äkkikuolemalle ja geneettiseen testaukseen synnynnäisen pitkän QT-oireyhtymän osalta. Mayo Clin Proc (2003) 78: 1479-87. doi:10.4065/78.12.1479
PubMed Abstrakti / Pubmed kokoteksti | CrossRef kokoteksti
42. Crotti L, Lundquist AL, Insolia R, Pedrazzini M, Ferrandi C, De Ferrari GM, et al. KCNH2-K897T on latentin synnynnäisen pitkän QT-oireyhtymän geneettinen muunnos. Levikki (2005) 112:1251-8. doi: 10.1161 / CIRCULATIONAHA.105.549071
Pubmed Abstrakti / Pubmed Koko Teksti | CrossRef Koko Teksti
43. Nof E, Cordeiro JM, Pérez GJ, Scornik FS, Calloe K, Love B ym. Tavallinen yksittäinen nukleotidipolymorfismi voi pahentaa kätkytkuolemaan johtavaa pitkän QT-ajan tyypin 2 oireyhtymää. Circ Cardiovasc Genet (2010) 3:199-206. doi: 10.1161 / SIRKGENETIIKKA.109. 898569
Pubmed Abstrakti / Pubmed Koko Teksti | CrossRef Koko Teksti
44. Bezzina C, Veldkamp MW, van Den Berg MP, Postma AV, Rook MB, Viersma JW, et al. Yksittäinen Na (+)-kanavamutaatio, joka aiheuttaa sekä pitkän QT-että Brugada-oireyhtymiä. Circ Res (1999) 85: 1206-13. doi: 10.1161 / 01.RES.85.12.1206
PubMed Abstrakti / Pubmed kokoteksti | CrossRef kokoteksti
45. Kyndt F, Probst V, Potet F, Demolombe S, Chevallier JC, Baro I, et al. Uusi SCN5A-mutaatio, joka johtaa joko eristettyyn sydämen johtumishäiriöön tai Brugadan oireyhtymään suuressa ranskalaisperheessä. Levikki (2001) 104:3081-6. doi: 10.1161 / hc5001.100834
Pubmed Abstrakti / Pubmed koko teksti | CrossRef koko teksti
46. Makita N, Behr E, Shimizu W, Horie M, Sunami A, Crotti L, et al. SCN5A: n e1784k-mutaatioon liittyy tyypin 3 pitkän QT-oireyhtymän sekalainen kliininen fenotyyppi. J Clin Invest (2008) 118:2219-29. doi: 10.1172 / JCI34057
Pubmed Abstrakti / Pubmed kokoteksti | CrossRef kokoteksti
47. Keller DI, Acharfi s, Delacrétaz E, Benammar N, Rotter M, Pfammatter JP, et al. Uusi mutaatio SCN5A: ssa, delQKP 1507-1509, aiheuttaen pitkän QT-oireyhtymän: q1507-jäännöksen rooli natriumkanavan inaktivaatiossa. J Mol Cell Cardiol (2003) 35: 1513-21. doi: 10.1016 / j.yjmcc.2003.08.007
Pubmed Abstrakti / Pubmed Koko Teksti | CrossRef Koko Teksti
48. Priori SG, Schwartz PJ, Napolitano C, Bloise R, Ronchetti E, Grillo m, et al. Riskiositus pitkän QT-ajan oireyhtymässä. N Engl J Med (2003) 348: 1866-74. doi: 10.1056 / NEJMoa022147
Pubmed Abstrakti / Pubmed koko teksti | CrossRef koko teksti
49. Lupoglazoff JM, Cheav T, Baroudi G, Berthet M, Denjoy I, Cauchemez B, et al. Homotsygoottinen SCN5A-mutaatio pitkän QT-ajan oireyhtymässä, jossa on toiminnallinen kahden suhde yhteen eteis-kammiokatkos. Circ Res (2001) 89: E16–21. doi: 10.1161 / hh1401. 095087
CrossRef koko teksti
50. Shi R, Zhang Y, Yang C, Huang C, Zhou X, Qiang h, et al. Sydämen natriumkanavamutaatio delQKP 1507-1509 liittyy lqt3: n laajenevaan fenotyyppiseen spektriin, johtumishäiriöön, laajentuneeseen kardiomyopatiaan ja korkeaan nuorten äkkikuolemien esiintyvyyteen. Europace (2008) 10:1329-35. doi: 10.1093 / europace / eun202
Pubmed Abstrakti / Pubmed koko teksti | CrossRef koko teksti
51. Zumhagen S, Veldkamp MW, Stallmeyer B, Baartsche a, Eckardt L, Paul M, et al. Sydämen natriumkanavageenin SCN5A: n heterotsygoottinen deleetiomutaatio, jolla on toimintakyvyn menetys – ja vahvistumisominaisuuksia, ilmenee eristettynä johtumissairautena, ilman merkkejä Brugada-oireyhtymästä tai pitkän QT-ajan oireyhtymästä. PLoS One (2013) 8:e67963. doi: 10.1371 / lehti.pone.0067963
Pubmed Abstrakti / Pubmed Koko Teksti | CrossRef Koko Teksti
52. Tuotantolaitos LD, Bowers PN, Liu Q, Morgan t, Zhang T, State MW, et al. A common Heart sodium channel variant associated with sudden infant death in African Americans, SCN5A S1103Y.J Clin Invest (2006) 116:430-5. doi: 10.1172 / JCI25618
Pubmed Abstrakti / Pubmed kokoteksti | CrossRef kokoteksti
53. Mohler PJ, Schott JJ, Gramolini AO, Dilly KW, Guatimosim s, duBell WH, et al. Ankyriini-B-mutaatio aiheuttaa tyypin 4 pitkän QT-ajan sydämen rytmihäiriöitä ja sydänperäisen äkkikuoleman. Nature (2003) 421: 634-9. doi: 10.1038 / nature01335
Pubmed Abstrakti / Pubmed kokoteksti | CrossRef kokoteksti
54. Schott JJ, Charpentier F, Peltier s, Foley P, Drouin E, Bouhour JB, et al. Pitkän QT-syndrooman geenin kartoittaminen kromosomiin 4q25-27. Am J Hum Genet (1995) 57:1114-22.
Pubmed Abstrakti / Pubmed Kokoteksti
55. Barhanin J, Lesage F, Guillemare E, Fink M, Lazdunski M, Romey G. K(V)LQT1 ja LSK (minkki) proteiinit liittyvät muodostaen I(Ks) – sydämen kaliumvirran. Nature (1996) 384: 78-80. doi: 10.1038 / 384078a0
Pubmed Abstrakti / Pubmed kokoteksti | CrossRef kokoteksti
56. Sanguinetti MC, Curran ME, Zou A, Shen J, Spector PS, Atkinson DL, et al. K(V)LQT1-ja minkin (IsK) proteiinien koossapysyminen sydämen I(Ks) – kaliumkanavan muodostamiseksi. Nature (1996) 384: 80-3. doi: 10.1038 / 384080a0
Pubmed Abstrakti / Pubmed koko teksti | CrossRef koko teksti
57. Schulze-Bahr E, Wang Q, Wedekind H, Haverkamp W, Chen Q, Sun Y, et al. KCNE1-mutaatiot aiheuttavat Jervellin ja Lange-Nielsenin oireyhtymää. Nat Genet (1997) 17:267-8. doi: 10.1038/ng1197-267
CrossRef koko teksti
58. Duggal P, Vesely MR, Wattanasirichaigoon D, Villafane J, Kaushik V, Beggs AH. Geenin mutaatio IK: ssa liittyy sekä Jervellin että Lange-Nielsenin ja Romano-Wardin pitkän QT-oireyhtymän muotoihin. Levikki (1998) 97: 142-6. doi: 10.1186/1471-2350-9-24
PubMed Abstrakti / Pubmed Kokoteksti | CrossRef Kokoteksti
59. Nishio Y, Makiyama T, Itoh H, Sakaguchi T, Ohno S, Gong YZ, et al. D85N, kcne1-polymorfismi, on pitkän QT-oireyhtymän sairautta aiheuttava geenimuunnos. J Am Coll Cardiol (2009) 54:812-9. doi: 10.1016 / J.jacc.2009. 06. 005
Pubmed Abstrakti | Pubmed Koko Teksti | CrossRef Koko Teksti
60. Paulussen AD, Gilissen RA, Armstrong M, Doevendans PA, Verhasselt P, Smeets HJ, et al. Kcnq1: n, KCNH2: n, SCN5A: n, KCNE1: n ja kcne2: n geneettiset vaihtelut lääkkeiden indusoimaa pitkää QT-oireyhtymää sairastavilla potilailla. Mol Med (2004) 82: 182-8. doi: 10.1007 / s00109-003-0522-z
Pubmed Abstrakti / Pubmed koko teksti | CrossRef koko teksti
61. Abbott GW, Sesti F, Splawski I, Buck ME, Lehmann MH, Timothy KW, et al. MiRP1 muodostaa Hergin kanssa ikr-kaliumkanavia ja siihen liittyy sydämen rytmihäiriöitä. Cell (1999) 97:175-87. doi: 10.1016 / S0092-8674 (00)80728-X
Pubmed Abstrakti / Pubmed kokoteksti | CrossRef kokoteksti
62. Gordon E, Panaghie G, Deng L, Bee KJ, Roepke TK, Krogh-Madsen T, et al. Kcne2-mutaatio potilaalla, jolla on kuuloärsykkeiden ja seerumin elektrolyyttitasapainon epätasapainon aiheuttama sydämen rytmihäiriö. Cardiovasc Res (2008) 77:98-106. doi: 10.1093 / cvr/cvm030
Pubmed Abstrakti / Pubmed kokoteksti | CrossRef kokoteksti
63. Plaster NM, Tawil R, Tristani-Firouzi M, Canún s, Bendahhou s, Tsunoda A, et al. Kir2.1: n mutaatiot aiheuttavat Andersenin oireyhtymän kehityshäiriöitä ja episodisia sähköisiä fenotyyppejä. Cell (2001) 105:511-9. doi: 10.1016 / S0092-8674(01)00342-7
PubMed Abstrakti / Pubmed Kokoteksti | CrossRef Kokoteksti
64. Kubo Y, Baldwin TJ, Jan YN, Jan LY. Ensisijainen rakenne ja toiminnallinen ilmentymä hiiren sisäänpäin tasasuuntaaja kaliumkanava. Nature (1993) 362:127-33. doi: 10.1038 / 362127a0
Pubmed Abstrakti / Pubmed koko teksti | CrossRef koko teksti
65. Raab-Graham KF, Radeke CM, Vandenberg CA. Molekyyli Kloonaus ja ilmentymä ihmissydämen sisäänpäin tasasuuntaaja kalium kanava. Neuroraportti (1994) 5: 2501-5. doi: 10.1097/00001756-199412000-00024
PubMed Abstrakti / Pubmed Kokoteksti | CrossRef Kokoteksti
66. Splawski I, Timothy KW, Sharpe LM, Decher N, Kumar P, Bloise R, et al. Ca(V)1.2 kalsiumkanavan toimintahäiriö aiheuttaa monijärjestelmän häiriötä, kuten rytmihäiriöitä ja autismia. Cell (2004) 119:19-31. doi: 10.1016 / J.solu.2004. 09. 011
Pubmed Abstrakti | Pubmed Koko Teksti | CrossRef Koko Teksti
67. Splawski I, Timothy KW, Decher N, Kumar P, Sachse FB, Beggs AH, et al. Sydämen L-tyypin kalsiumkanavamutaatioiden aiheuttama vaikea rytmihäiriöhäiriö. Proc Natl Acad Sci U S A (2005) 102:8089-96. doi: 10.1073 / pnas.0502506102
Pubmed Abstrakti / Pubmed Koko Teksti | CrossRef Koko Teksti
68. Gillis J, Burashnikov E, Antzelevitch C, Blaser S, Gross G, Turner L, et al. Pitkä QT, syndactyly, nivelkontraktuurit, aivohalvaus ja uusi Cacna1c-mutaatio: Timothyn oireyhtymän spektrin laajentaminen. Am J Med Genet A (2012) 158A:182-7. doi: 10.1002 / ajmg.a.34355
Pubmed Abstrakti / Pubmed Koko Teksti | CrossRef Koko Teksti
69. Dufendach KA, Giudicessi JR, Boczek NJ, Ackerman MJ. Äidin mosaiikkia hämmentää vastasyntyneen diagnoosi tyypin 1 Timothy oireyhtymä. Pediatrics (2013) 131:e1991–5. doi: 10.1542 / peds.2012-2941
Pubmed Abstrakti / Pubmed Kokoteksti | CrossRef Kokoteksti
70. Vatta M, Ackerman MJ, Ye B, Makielski JC, Ughanze EE, Taylor EW, et al. Mutantti caveolin – 3 indusoi pysyvää myöhäisnatriumvirtaa ja siihen liittyy pitkän QT-ajan oireyhtymä. Levikki (2006) 114:2104–12. doi: 10.1161 / CIRCULATIONAHA.106. 635268
Pubmed Abstrakti / Pubmed Koko Teksti | CrossRef Koko Teksti
71. Cronk LB, Ye B, Kaku T, Tester DJ, Vatta M, Makielski JC, et al. Novel mechanism for sudden infant death syndrome: persistent late sodium current secondary to mutations in caveolin-3. Heart Rhythm (2007) 4:161-6. doi: 10.1016 / J. hrthm.2006. 11. 030
Pubmed Abstrakti | Pubmed Koko Teksti | CrossRef Koko Teksti
72. Engelman JA, Zhang X, Galbiati F, Volonte D, Sotgia F, Pestell RG, et al. Molecular genetics of the caveolin gene family: implications for human cancer, diabetes, Alzheimer disease, and muscular dystrofy. Am J Hum Genet (1998) 63:1578-87. doi:10.1086/302172
yliviivataan koko teksti
73. Balijepalli RC, Foell JD, Hall DD, Hell JW, Kamp TJ. Sydämen L-tyypin Ca(2+)-kanavien lokalisointi caveolaariseen makromolekyyliseen signalointikompleksiin tarvitaan beeta(2) – adrenergiseen säätelyyn. Proc Natl Acad Sci U S A (2006) 103:7500-5. doi: 10.1073 / pnas.0503465103
Pubmed Abstrakti / Pubmed Koko Teksti | CrossRef Koko Teksti
74. Medeiros-Domingo A, Kaku T, Tester DJ, Iturralde-Torres P, Itty A, Ye B, et al. SCN4B-koodattu natriumkanava beta4-alayksikkö synnynnäisessä pitkän QT-oireyhtymän hoidossa. Levikki (2007) 116:134-42. doi: 10.1161 / CIRCULATIONAHA.106. 659086
Pubmed Abstrakti / Pubmed Koko Teksti | CrossRef Koko Teksti
75. Chen L, Marquardt ML, Tester DJ, Sampson KJ, Ackerman MJ, Kasss RS. A-kinaasia ankkuroivan proteiinin mutaatio aiheuttaa pitkän QT-ajan oireyhtymän. Proc Natl Acad Sci U S A (2007) 104:20990-5. doi: 10.1073 / pnas.0710527105
Pubmed Abstrakti / Pubmed Koko Teksti | CrossRef Koko Teksti
76. Wu G, Ai T, Kim JJ, Mohapatra B, Xi Y, Li Z, et al. Alfa-1-syntrofiinimutaatio ja pitkä QT-oireyhtymä: natriumkanavahäiriöiden sairaus. Circ Arrythm Electrophysiol (2008) 1:193-201. doi: 10.1161 / CIRCEP.108. 769224
Pubmed Abstrakti / Pubmed Koko Teksti | CrossRef Koko Teksti
77. Yang Y, Yang Y, Liang B, Liu J, Li J, Grunnet m, et al. Kir3: n tunnistus.4 mutaatio synnynnäisessä pitkän QT-ajan oireyhtymässä. Am J Hum Genet (2010) 86:872-80. doi: 10.1016 / J.ajhg.2010. 04. 017
Pubmed Abstrakti | Pubmed Kokoteksti | CrossRef Kokoteksti
78. Roden DM. Proarytmian mekanismit ja hallinta. Am J Cardiol (1998) 82: 49I-57I. doi: 10.1016 / S0002-9149 (98)00472-X
CrossRef koko teksti
79. Mitcheson JS, Chen J, Lin M, Culberson C, Sanguinetti MC. Rakenteellinen perusta lääkkeiden aiheuttamalle pitkä QT-oireyhtymälle. Proc Natl Acad Sci U S A (2000) 97:12329-33. doi: 10.1073 / pnas.210244497
Risti Koko Teksti
80. Kannankeril PJ, Roden DM. Drug-induced long QT and torsade de pointes: recent advances. Curr Opin Cardiol (2007) 22:39-43. doi: 10.1097 / HCO.0b013e32801129eb
Pubmed Abstrakti / Pubmed koko teksti | CrossRef koko teksti
81. Veerman CC, Verkerk AO, Blom MT, Klemens CA, Langendijk PN, van Ginneken AC, et al. Hidas viivästynyt tasasuuntaajan kaliumvirtasalpaus vaikuttaa merkittävästi lääkkeen aiheuttamaan pitkän QT-oireyhtymän syntyyn. Circ Arrythm Electrophysiol (2013) 6:1002-9. doi: 10.1161 / CIRCEP.113.000239
Pubmed Abstrakti / Pubmed Koko Teksti | CrossRef Koko Teksti
82. Sesti F, Abbott GW, Wei J, Murray KT, Saksena S, Schwartz PJ, et al. Yleinen polymorfismi, joka liittyy antibioottien aiheuttamaan sydämen rytmihäiriöön. Proc Natl Acad Sci U S A (2000) 97:10613-8. doi: 10.1073 / pnas.180223197
Pubmed Abstrakti / Pubmed Koko Teksti | CrossRef Koko Teksti
83. Käärb s, Crawford DC, Sinner MF, Behr ER, Kannankeril PJ, Wilde AA, et al. Suuri ehdokas geenitutkimus tunnistaa kcne1 D85N polymorfismi mahdollisena modulaattori huumeiden aiheuttama kääntyvien kärkien. Circ Cardiovasc Genet (2012) 5:91-9. doi: 10.1161 / SIRKGENETIIKKA.111. 960930
Pubmed Abstrakti / Pubmed Koko Teksti | CrossRef Koko Teksti
84. Nakamura K, Katayama Y, Kusano KF, Haraoka K, Tani Y, Nagase s, et al. Anti-KCNH2 vasta-aineiden aiheuttama pitkä QT-oireyhtymä: Uusi hankittu muoto pitkä QT-oireyhtymä. J Am Coll Cardiol (2007) 50:1808-9. doi: 10.1016 / J.jacc.2007. 07. 037
CrossRef Koko Teksti
85. Moss AJ, Zareba W, Benhorin J, Locati EH, Hall WJ, Robinson JL, et al. EKG t-aaltomallit perinnöllisen pitkän QT-oireyhtymän geneettisesti erillisissä muodoissa. Levikki (1995) 92: 2929-34. doi: 10.1161 / 01.CIR.92. 10. 2929
Pubmed Abstrakti | Pubmed Koko Teksti | CrossRef Koko Teksti
86. Zhang l, Timothy KW, Vincent GM, Lehmann MH, Fox J, Giuli LC ym. St-t-aaltokuvioiden ja repolarisaatioparametrien spektri synnynnäisessä pitkän QT-oireyhtymän oireyhtymässä: EKG-löydökset tunnistavat genotyypit. Levikki (2000) 102:2849-55. doi: 10.1161 / 01.CIR.102.23.2849
Pubmed Abstrakti / Pubmed Koko Teksti | CrossRef Koko Teksti
87. Tan HL, Bardai A, Shimizu W, Moss AJ, Schulze-Bahr E, Noda T, et al. Genotyyppispesifinen rytmihäiriöiden puhkeaminen synnynnäisessä pitkän QT-oireyhtymän hoidossa: mahdolliset hoidon vaikutukset. Levikki (2006) 114:2096-103. doi: 10.1161 / CIRCULATIONAHA.106. 642694
Pubmed Abstrakti / Pubmed Koko Teksti | CrossRef Koko Teksti
88. Locati EH, Zareba W, Moss AJ, Schwartz PJ, Vincent GM, Lehmann MH, et al. Ikään ja sukupuoleen liittyvät erot kliinisissä ilmenemismuodoissa potilailla, joilla on synnynnäinen pitkän QT-ajan oireyhtymä: kansainvälisen LQTS-rekisterin löydökset. Levikki (1998) 97: 2237-44. doi: 10.1161 / 01.CIR.97. 22. 2237
Pubmed Abstrakti | Pubmed Koko Teksti | CrossRef Koko Teksti
89. Zareba W, Moss AJ, Schwartz PJ, Vincent GM, Robinson JL, Priori SG, et al. Genotyypin vaikutus pitkän QT-ajan oireyhtymän kliiniseen kulkuun. International Long-QT Syndrome Registry Research Group. N Engl J Med (1998) 339: 960-5. doi: 10.1056 / NEJM199810013391404
Pubmed Abstrakti / Pubmed koko teksti | CrossRef koko teksti
90. Goldenberg I, Moss AJ, Bradley J, Polonsky S, Peterson DR, McNitt s, et al. Pitkän QT-ajan oireyhtymä 40 ikävuoden jälkeen. Levikki (2008) 117:2192-201. doi: 10.1161 / CIRCULATIONAHA.107. 729368
Pubmed Abstrakti / Pubmed Koko Teksti | CrossRef Koko Teksti
91. Zareba W, Moss AJ, Locati EH, Lehmann MH, Peterson DR, Hall WJ, et al. Syndroomarekisteri. Iän ja sukupuolen vaikutusten modulointi pitkän QT-oireyhtymän kliiniseen kulkuun genotyypin mukaan. J Am Coll Cardiol (2003) 42:103-9. doi: 10.1016 / S0735-1097(03)00554-0
PubMed Abstrakti / Pubmed kokoteksti | CrossRef kokoteksti
92. Seth R, Moss AJ, McNitt S, Zareba W, Andrews ML, Qi m, et al. Pitkä QT-oireyhtymä ja raskaus. J Am Coll Cardiol (2007) 49:1092-8. doi: 10.1016 / J.jacc.2006. 09. 054
Pubmed Abstrakti | Pubmed Koko Teksti | CrossRef Koko Teksti
93. Schwartz PJ, Priori SG, Dumaine R, Napolitano C, Antzelevitch C, Stramba-Badiale m, et al. Molekyyliyhteys kätkytkuoleman ja pitkän QT: n välillä. N Engl J Med (2000) 343: 262-7. doi: 10.1056/NEJM200007273430405
CrossRef koko teksti
94. Schwartz PJ, Priori SG, Bloise R, Napolitano C, Ronchetti E, Piccinini A, et al. Molekyylidiagnoosi lapsella, jolla on kätkytkuolema. Lancet (2001) 358:1342-3. doi: 10.1016 / S0140-6736(01)06450-9
PubMed Abstrakti / Pubmed kokoteksti | CrossRef kokoteksti
95. Christiansen M, Tønder N, Larsen LA, Andersen PS, Simonsen H, Oyen N, et al. Mutations in the HERG K + – ion channel: a novel link between long QT syndrome and sudden infant death syndrome. Am J Cardiol (2005) 95:433-4. doi: 10.1016 / J.amjcard.2004. 09. 054
Pubmed Abstrakti | Pubmed Koko Teksti | CrossRef Koko Teksti
96. Testaaja DJ, Ackerman MJ. Kuolemanjälkeinen pitkä QT-syndrooma geneettinen tutkimus selittämättömän äkkikuoleman varalta nuorilla. J Am Coll Cardiol (2007) 49:240-6. doi: 10.1016 / J.jacc.2006. 10. 010
Pubmed Abstrakti | Pubmed Koko Teksti | CrossRef Koko Teksti
97. Hofman N, Tan HL, Clur SA, Alders M, van Langen IM, Wilde AA. Perinnöllisen sydänsairauden osuus sydänperäiseen äkkikuolemaan lapsuudessa. Pediatrics (2007) 120:e967–73. doi: 10.1542 / peds.2006-3751
Pubmed Abstrakti / Pubmed Koko Teksti | CrossRef Koko Teksti
98. Wedekind H, Smits JP, Schulze-Bahr E, Arnold R,Veldkamp MW, Bajanowski T, et al. De novo-mutaatio SCN5A-geenissä, joka liittyy varhaiseen kätkytkuolemaan. Levikki (2001) 104:1158-64. doi: 10.1161/hc3501.095361
CrossRef koko teksti
99. Wang DW, Desai RR, Crotti L, Arnestad M, Insolia R, Pedrazzini m, et al. Sydämen natriumkanavan toimintahäiriö kätkytkuoleman oireyhtymässä. Levikki (2007) 115:368-76. doi: 10.1161 / CIRCULATIONAHA.106. 646513
Pubmed Abstrakti / Pubmed Koko Teksti | CrossRef Koko Teksti
100. Van Norstrand DW, Valdivia CR, Tester DJ, Ueda K, London B, Makielski JC, et al. Uusien glyseroli-3-fosfaattidehydrogenaasi 1: n kaltaisten geenin (GPD1-L) mutaatioiden molekyyli-ja funktionaalinen Luonnehdinta kätkytkuoleman oireyhtymässä. Levikki (2007) 116:2253-9. doi: 10.1161 / CIRCULATIONAHA.107. 704627
Pubmed Abstrakti / Pubmed Koko Teksti | CrossRef Koko Teksti
101. Tan BH, Pundi KN, Van Norstrand DW, Valdivia CR, Tester DJ, Medeiros-Domingo a, et al. Sudden infant death syndrome-associated mutations in the sodium channel beta alayksiköt. Heart Rhythm (2010) 7: 771-8. doi: 10.1016 / J. hrthm.2010. 01. 032
Pubmed Abstrakti | Pubmed Kokoteksti | CrossRef Kokoteksti
102. Miller TE, Estrella E, Myerburg RJ, Garcia de Viera J, Moreno N, Rusconi P, et al. Toistuva kolmannen raskauskolmanneksen sikiön menetys ja äidin mosaiikki pitkän QT-oireyhtymän vuoksi. Levikki (2004) 109:3029-34. doi: 10.1161 / 01.CIR.0000130666. 81539. 9 E
Pubmed Abstrakti / Pubmed Kokoteksti | Poikkiteksti Kokoteksti
103. Chockalingam P, Crotti L, Girardengo G, Johnson JN, Harris KM, van der Heijden JF, et al. Kaikki beetasalpaajat eivät ole samanarvoisia pitkän QT-oireyhtymän tyyppien 1 ja 2 hoidossa: metoprololilla esiintyvien tapahtumien toistuvuus on suurempi. J Am Coll Cardiol (2012) 60:2092-6. doi: 10.1016 / J.jacc.2012.07.046
Pubmed Abstrakti / Pubmed Koko Teksti | CrossRef Koko Teksti
104. Schwartz PJ, Priori SG, Cerrone M,Spazzolini C, Odero A, Napolitano C, et al. Vasemman sydämen sympaattinen denervaatio korkean riskin pitkän QT-oireyhtymän hoidossa. Levikki (2004) 109:1826-33. doi: 10.1161 / 01.CIR.0000125523. 14403. 1 E
Pubmed Abstrakti / Pubmed Kokoteksti | Poikkiteksti Kokoteksti
105. Singh B, al Shahwan SA, Habbab MA, al Deeb SM, Biary N. idiopaattinen pitkä QT-oireyhtymä: oikean kysymyksen esittäminen. Lancet (1993) 341:741. doi:10.1016/0140-6736(93)90501-7
yliviivataan koko teksti
106. Gorgels AP, Al Fadley F, Zaman L, Kantoch MJ, Al Halees Z. pitkä QT-oireyhtymä, jolla on heikentynyt eteis-kammiojohtuminen: pahanlaatuinen variantti imeväisillä. J Cardiovasc Electrophysiol (1998) 9:1225-32. doi: 10.1111 / j.1540-8167. 1998.tb00096.x
Pubmed Abstrakti / Pubmed koko teksti | CrossRef koko teksti
107. Kantoch MJ, Qurashi MM, Bulbul ZR, Gorgels AP. Vastasyntynyt, jolla on monimutkainen synnynnäinen sydänsairaus, eteis-kammiokatkos ja kääntyvien kärkien kammiotakykardia. Pacing Clin Electrophysiol (1998) 21:2664-7. doi: 10.1111 / j.1540-8159. 1998.tb00043.x
CrossRef koko teksti
108. El-Hazmi MA, al-Swailem AR, Warsy AS, al-Swailem AM, Sulaimani R, al-Meshari AA. Verisukulaisuus Saudi-Arabian väestön keskuudessa. J Med Genet (1995) 32:623-6. doi: 10.1136 / jmg.32. 8. 623
CrossRef Koko Teksti
109. El Mouzan MI, Al Salloum AA, Al Herbish AS, Qurachi MM, Al Omar AA. Consangulinity and major genetic disorders in Saudi children: a community-based cross-sectional study. Ann Saudi Med (2008) 28:169-73. doi:10.4103/0256-4947.51726
PubMed Abstrakti / Pubmed kokoteksti | CrossRef kokoteksti
110. Medeiros-Domingo A, Bhuiyan ZA, Tester DJ, Hofman N, Biker H, van Tintelen JP, et al. RYR2-koodattu ryanodiinireseptori / kalsiumin vapautuskanava potilailla, joilla on aiemmin todettu joko katekoliaminerginen polymorfinen kammiotakykardia tai genotyyppinegatiivinen, liikunnan aiheuttama pitkän QT-ajan oireyhtymä: kattava avoin lukukehys mutaatioanalyysi. J Am Coll Cardiol (2009) 54:2065-74. doi: 10.1016 / J.jacc.2009. 08. 022
Pubmed Abstrakti | Pubmed Koko Teksti | CrossRef Koko Teksti
111. Al-Aama JY, Al-Ghamdi S, Bdier AY, Wilde AA, Bhuiyan ZA. De novo-mutaatio Kcnq1-geenissä aiheuttaa Jervellin ja Lange-Nielsenin oireyhtymän. Clin Genet (2013). doi: 10.1111 / cge.12300
Pubmed Abstrakti / Pubmed Koko Teksti | CrossRef Koko Teksti
112. Splawski I, Timothy KW, Tateyama M, Clancy CE, Malhotra A, Beggs AH, et al. SCN5A – natriumkanavan muunnos, joka liittyy sydämen rytmihäiriöriskiin. Tiede (2002) 297:1333-6. doi: 10.1126 / tiede.1073569
CrossRef Koko Teksti
113. Wilde AA, Bezzina CR. Sydämen rytmihäiriöiden genetiikka. Sydän (2005) 91: 1352-8.