Huuli-ja kitalakihalkion
 Intro/abstractCleft-huulen genetiikka, jossa on tai ei ole suulakihalkiota, on monimutkainen synnynnäinen poikkeama, joka voidaan eristää tai nähdä yhdessä muiden epämuodostumien kanssa. Se voi olla myös osa geneettisen oireyhtymän fenotyyppiä. Tässä artikkelissa tarkastellaan huuli-ja suulakihalkion yleisyyttä, uusiutumisriskejä ja muiden synnynnäisten poikkeavuuksien riskejä. Tutkitaan geneettisiä oireyhtymiä ja teratogeenisiä altistuksia, joiden tiedetään liittyvän suun halkeamiin. Lisäksi keskustellaan geneettisistä testeistä, joita yleisesti pyydetään pediatric clinical genetics-ympäristössä huuli-ja suulakihalkioisen potilaan arvioimiseksi.
Intro/abstractCleft-huulen genetiikka, jossa on tai ei ole suulakihalkiota, on monimutkainen synnynnäinen poikkeama, joka voidaan eristää tai nähdä yhdessä muiden epämuodostumien kanssa. Se voi olla myös osa geneettisen oireyhtymän fenotyyppiä. Tässä artikkelissa tarkastellaan huuli-ja suulakihalkion yleisyyttä, uusiutumisriskejä ja muiden synnynnäisten poikkeavuuksien riskejä. Tutkitaan geneettisiä oireyhtymiä ja teratogeenisiä altistuksia, joiden tiedetään liittyvän suun halkeamiin. Lisäksi keskustellaan geneettisistä testeistä, joita yleisesti pyydetään pediatric clinical genetics-ympäristössä huuli-ja suulakihalkioisen potilaan arvioimiseksi.
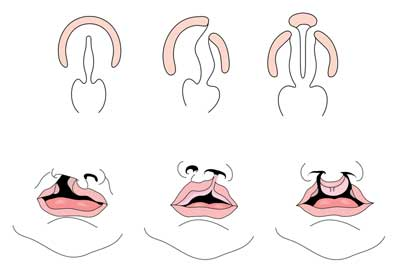
huulihalkio suulakihalkiolla tai ilman suulakihalkiota (CL/CP) eroaa eristetystä suulakihalkiosta alkion, epidemiologisen ja geneettisen tason perusteella. Halkeama huuli tyypillisesti johtuu yläleuan näkyvyyttä ja mediaalinen nenän näkyvyyttä ei sulake välillä viidennen ja kuudennen viikon alkion kehitystä. Normaali kitalaen kehitys johtuu primaarisen kitalaen ja sekundaarisen kitalaen muodostumisesta. Ensisijainen kitalaki muodostuu viikoilla kuudesta seitsemään kehittämällä ja fuusio mediaalinen nenän, lateral nenän, ja yläleuan prosesseja. Toissijainen kitalaki on peräisin palataalihyllyistä (jotka kehittyvät ensimmäisen haarakaaren parillaalisista prosesseista), jotka tulevat vaakasuoriksi ja sulautuvat muodostaen kovat ja pehmeät palat noin yhdeksännellä alkionkehitysviikolla. Hyllyt sulautuvat myös ensisijaiseen kitalakeen ja nenän väliseinään. (1)
suun halkeamat ovat yksi yleisimmistä vastasyntyneiden lasten sikiövaurioista, ja niiden yleisyys on 1.6 tuhatta vastasyntynyttä kohti maailmanlaajuisesti, CL / CP nähdään noin yhdessä tuhannessa synnytyksessä ja CP nähdään 0,6 tuhannessa synnytyksessä. (2) CL/CP on yleisempää aasialaisilla, afrikkalaisilla ja Alkuperäisamerikkalaisilla. CL / CP on myös yleisempi miehillä. Sen sijaan CP: n esiintyvyydessä ei ole merkittävää eroa eri etnisten taustojen välillä, ja CP on yleisempää naisilla. (3) uusiutumisriskit perheessä riippuvat siitä, onko halkeama eristetty (ilman muita kliinisiä löydöksiä) vai onko se osa geneettistä oireyhtymää. Useimmat oraaliset halkeamat ovat yksittäisiä (noin 80%). Eristetyt halkeamat uskotaan olevan monitekijäinen perintö: ne johtuvat yhdistelmä useita tekijöitä, sekä geneettinen ja ympäristön. Uusiutumisriski (Taulukko 1) kasvaa, kun sairastuneita omaisia on useampi kuin yksi. Myös uusimisriski kasvaa, mitä vakavammasta viasta on kyse.

huuli-ja kitalakihalkio voi aiheuttaa muita synnynnäisiä anomalioita. Geneettisen tai teratogeenisen etiologian todennäköisyys kasvaa sitä enemmän, mitä enemmän potilaalla on synnynnäisiä poikkeavuuksia. Läsnäolo muita kysymyksiä, kuten kehitysvammaisuus, käyttäytymisen ongelmia, kuten autismi, dysmorphic ominaisuuksia, tai muita lääketieteellisiä huolenaiheita tekee myös geneettinen häiriö tai teratogeeninen altistuminen todennäköisempää. Noin 13% yksilöiden halkio huuli on muita lääketieteellisiä huolenaiheita tai poikkeavuuksia. Huuli-ja suulakihalkiolla määrä kasvaa 37 prosenttiin ja pelkällä suulakihalkiolla 47 prosenttiin.
Äitiysaltistuksen teratogeenisille aineille (kuten talidomidille, epilepsialääkkeille, alkoholille, retinoiinihapolle ja savukkeille) ja äidin sairauksien (kuten diabetes, vihurirokko ja folaatin puutos) on osoitettu lisäävän suun halkeamien riskiä. Myös lapsivesinauhojen esiintyminen lisää halkeamien riskiä. Periconceptual foolihappolisän tiedetään vähentävän suun halkeamien riskiä.
Pierre Robin sequence on kraniofacial anomalia ominaista alaleuan hypoplasia tai mikrognathia, toissijainen U-muotoinen suulakihalkio, ja glossoptosis johtaa obstruktiivinen uniapnea ja ruokinta vaikeuksia. Pierre Robinin sekvenssi voidaan nähdä osana geneettisiä oireyhtymiä (22q11.2 deleetio-oireyhtymä, Sticklerin oireyhtymä; kuvattu alla). (5)
suun halkeamiin liittyy satoja geneettisiä oireyhtymiä, mukaan lukien sytogeneettiset poikkeavuudet (aneuploidiat, mikrodeleetiot) ja yhden geenin (Mendelin) häiriöt. Geenidiagnoosin vahvistaminen on välttämätöntä ennusteen määrittämiseksi ja uusiutumisriskin määrittämiseksi.
Aneuploidit, kuten trisomia 13 ja 18, ovat vahvasti yhteydessä CL/CP: hen. Trisomia 13 (eli Pataun syndrooma) liittyy kromosomi 13: n kolmeen kopioon eli epätasapainoisiin Robertsonialaisiin translokaatioihin, joihin liittyy kromosomi 13. Vauvat syntyvät tämän ehdon tyypillisesti kuolee vastasyntyneen aikana. Kliinisiä piirteitä ovat huuli-ja suulakihalkio, kasvun hidastuminen, vakavat keskushermoston epämuodostumat (mukaan lukien holoprosenkefalia), mikrokefalia, mikrotalmia, värikalvon kolobooma, silmien puuttuminen, epämuodostuneet korvat, polydaktylia, nyrkkien puristuminen, keinujalka, synnynnäiset sydänviat ja urogenitaalivirheet. Trisomy 13: ssa voi esiintyä keskiviivan halkeamia (muuten hyvin harvinaisia) keskiviivan vikojen, kuten holoprosenkefalian, riskin vuoksi. Trisomia 18 (eli Edwardsin syndrooma) johtuu tyypillisesti kromosomi 18: n kolmesta erillisestä kopiosta, ja siihen liittyy huono postnataalinen tulos. Kliinisiä ominaisuuksia ovat huulihalkio ja kitalaki, kehitysvammaisuus, epäonnistuminen menestyä, synnynnäinen sydänsairaus, hypertonia, mikrognathia, lyhyt rintalasta, Alhainen asettaa epämuodostuneet korvat, puristetut kädet, keinu pohja jalat, ja hypoplastinen kynnet, muun muassa. Trisomia 13 ja 18 voidaan helposti vahvistaa tai sulkea pois tekemällä kromosomianalyysi (karyotyypin määritys).
Mikrodeletionioireyhtymissä on tyypillisesti kyse kromosomin osan poistamisesta. Nämä deleetiot voivat olla liian pieniä, jotta ne voitaisiin havaita tavanomaisella karyotyypin määrityksellä, ja ne voivat edellyttää kalojen havaitsemista (fluoresenssi in situ-hybridisaatio) tai mikroarray-tekniikkaa. Tunnettu mikrodeletion oireyhtymä liittyy suulakihalkio on 22q11.2 deleetio-oireyhtymä (alias Digeorge/Velocardiofacial syndrome). Palatal poikkeavuuksia kuten velopharyngeal epäpätevyys, submucosal clefts, bifid uvula, ja suulakihalkio nähdään 69% yksilöiden 22q11.2 poisto, ja voi olla osa Pierre Robin sekvenssi. Muita kliinisiä löydöksiä ovat synnynnäinen sydänsairaus, kuulon heikkeneminen, dysmorfiset piirteet, immuunipuutos, hypokalsemia, munuaispoikkeamat, ruokinta-asiat, luuston poikkeavuudet ja psykiatriset häiriöt. Noin 10% tapauksista 22q11.2 deleetio oireyhtymä uskotaan olevan familiaalinen. Poisto erottelee autosomaalisesti dominoivalla tavalla.(6) Wolf-Hirschhornin oireyhtymä, joka johtuu kromosomin 4 lyhyen varren deleetiosta, liittyy myös suun halkeamiin (25-50%: lla sairastuneista). Luonteenomaiset kasvonpiirteet (mukaan lukien näkyvä glabella, joka johtaa “kreikkalais-soturikypärän ulkonäköön”), synnynnäinen sydänsairaus, kehitysvammaisuus, kouristukset, epäonnistuminen menestyä, mikrognathia, preauricular tageja tai kuoppia, ja hypodontia voidaan myös nähdä osana kunnossa.(7)
Yksigeenisiä oraalihalkioisia häiriöitä ovat muun muassa Sticklerin oireyhtymä, Treacher Collinsin oireyhtymä ja Van der Wouden oireyhtymä. Sticklerin oireyhtymä on autosomissa dominoiva ja harvemmin autosomissa resessiivisesti periytyvä kollageenisairaus. Yhteisiä piirteitä ovat suulakihalkio (nähdään osana Pierre Robin sekvenssi tai ilman mikrognathia), kuulon heikkeneminen (sensorineuraalinen ja johtava), luuston löydökset (alussa niveltulehdus, spondyloepiphyseal dysplasia), silmien poikkeavuudet (korkea likinäköisyys, lasiaisen poikkeavuuksia) ja ominaiset kasvonpiirteet (alikehittyneisyys yläleuan ja nenän silta, midface retrusion). Sticklerin oireyhtymän geneettinen testaus voi olla monimutkaista, sillä sairastuneilla henkilöillä on kuvattu mutaatioita ainakin kuudessa geenissä. Noin 90% potilaista Stickler oireyhtymä on mutaatioita COL2A1 geeni ja on autosomaalinen hallitseva muoto ehto.(8) Treacher Collinsin oireyhtymä on autosomaalinen dominoiva tila, jolle on ominaista suulakihalkio huulihalkiolla tai ilman sitä 28%: lla sairastuneista henkilöistä. Muita poikkeavuuksia ovat poskiluiden ja alaleuan hypoplasia, ulkokorvan poikkeavuudet, alaluomen kolobooma, johtava kuulon heikkeneminen, alaripsien puuttuminen, preaurikulaaristen hiusten siirtyminen poskille ja choanal stenoosi tai atresia. Treacher Collinsin oireyhtymän diagnoosi perustuu kliinisiin ja radiologisiin löydöksiin. Mutaatioita on kuvattu ainakin kolmessa geenissä, ja TCOF1-mutaatioita on havaittu 78 – 93%: lla potilaista.(9) Van der Woude oireyhtymä on ominaista läsnäolo synnynnäinen, yleensä kahdenvälinen, paramedian alahuulen fistulae (kuoppia), tai joskus pieniä kumpuja sinus-suolikanavan johtaa limakalvojen rauhanen huulen, ja suun halkeamat (mukaan lukien CL/CP ja CP). Van der Woude on autosomaalinen dominoiva tila, joka liittyy mutaatioihin IRF6-geenissä (10). Yhden geenin tai monigeenin testaus edellyttää geenin suoraa analysointia sekvensoimalla ja/tai deleetio – / monistusanalyysillä (kuten MLPA).
koska geneettisiin oireyhtymiin, joihin liittyy huuli-ja suulakihalkio, voi liittyä aneuploidioita, kromosomin mikrodelektioita/mikroduplikaatioita tai yhden geenin häiriöitä, geneettinen testaus voi olla monimutkainen prosessi. Perusteellinen sairaushistoria, kolmen sukupolven sukutaulu, raskaushistoria ja dysmorphology tentti kliinisen geneetikon voi selventää kliinisen kuvan ja mahdollistaa kohdennetun geneettisen testauksen. Uudemmat teknologiat, mukaan lukien mikroarray, mahdollistavat pienten mikroelementtien ja mikroduplicaatioiden tunnistamisen, jotka aiemmin jäivät tavanomaisella karyotyypityksellä. Valitettavasti tämä tekniikka johtaa myös sellaisten poistojen ja päällekkäisyyksien tunnistamiseen, joilla ei ole kliinistä merkitystä, mikä vaikeuttaa geneettistä neuvontaprosessia. Yhden geenin häiriöiden tai Mendelin häiriöiden testaus edellyttää halutun geenin geneettisen testauksen kliinistä saatavuutta. Se voi myös olla kallista, jos ei kuulu sairausvakuutus. Uusia teknologioita, kuten seuraavan sukupolven sekvensointi, exome sekvensointi, tai genomin sekvensointi (tunnetaan yhdessä genomitestit) on nyt tullut kliinisesti saatavilla. Analysoimalla satoja tuhansia geenejä samanaikaisesti, nämä testit lisäävät merkittävästi diagnostista tehoa ja saantoa. Muihin tekniikoihin verrattuna nämä testit voivat antaa vastauksen nopeammin ja kustannustehokkaammin. Tutkimusalalla eksome-ja genomisekvensointi on johtanut uusien geenien tunnistamiseen sekä geneettisten mutaatioiden kliinisten ominaisuuksien ja spektrin laajenemiseen. Kuten microarray-tekniikalla, genomitesteillä voidaan havaita oireyhtymiä, jotka eivät liity potilaan esitystapaan ja/tai testauksen syyhyn. Geneettisen testauksen luontaisen monimutkaisuuden vuoksi tietoon perustuva suostumus on tarpeen.
johtopäätös
vaikka huuli-ja suulakihalkio on useimmissa tapauksissa yksittäinen poikkeama, suun halkeamien ja muiden poikkeavuuksien ja geneettisten oireyhtymien välillä on vahva yhteys. Kliinisen geneetikon ja geenineuvojan tekemä geeniarvio on välttämätön ennakoivan ohjauksen ja uusiutumisriskien määrittämiseksi. Geneettistä testausta, joka edellyttää tietoon perustuvaa suostumusta, voidaan koordinoida ja tulkita geneettisen arvioinnin aikana.
Anya Revah, MS, on johtava geneettinen neuvonantaja lääketieteellisen genetiikan osastolla Maimonides Infants and Children ‘ s Hospitalissa Brooklynissa, New Yorkissa. Hän on myös aktiivinen jäsen Maimonides Medical Center ja Kings County Hospital huuli-ja suulakihalkio monitieteinen joukkue. Hänellä on maisterintutkinto Geenineuvonnasta Bostonin yliopistosta Bostonissa, Massachusettsissa.
1. Sadler TW. Langmanin lääketieteellinen embryologia. Yhdeksäs Painos. Sivut 390-395.
2. Parker SE, Mai CT, Canfield MA, Rickard R, Wang Y, Meyer RE, Anderson P, Mason CA, Collins JS, Kirby RS, Correa A. kansallisen Syntymävaurioiden Ehkäisyverkko. Päivitetty kansallisen syntyvyyden yleisyysarviot valituille sikiövaurioille Yhdysvalloissa. 2004-2006. Birth Defects Research (Part A): Clinical and Molecular Teratology 2010;88: 1008-1016.
3. Fraser FC. Huulihalkion ja suulakihalkion genetiikka. On. J. Hum. Genet. 1970;22: 336–352.
4. Van Rooij IA, Ocke MC, et al. Periconceptual folate saanti täydentää ja elintarvikkeiden saanti vähentää riskiä ei-oireyhtymä huulihalkio tai ilman suulakihalkio. Prev Med 2004; 39: 689-694.
5. Tan TY. Kilpatrick N, Farlie PG. Kehitys-ja geneettisiä näkökulmia Pierre Robinin sekvenssiin. On. J. Med. Genet. 2013;163C:295-305.
6. McDonald-McGinn DM, Emanuel BS, ZACKAI EH. 22q11.2 deleetio-oireyhtymä. Syyskuuta. 23, 1999. . Julkaisussa: Pagon RA, Adam MP, Ardinger HH, et al., muokkaus. Yleisnäkemyksiä . Seattle (WA): University of Washington, Seattle; 1993-2014. Saatavilla: http://www.ncbi.nlm.nih.gov/books/NBK1523/.
7. Battaglia A, Carey JC, South ST, et al. Wolf-Hirschhornin Syndrooma. Huhti. 29, 2002. . Julkaisussa: Pagon RA, Adam MP, Ardinger HH, et al., muokkaus. Yleisnäkemyksiä . Seattle (WA): University of Washington, Seattle; 1993-2014. Saatavilla: http://www.ncbi.nlm.nih.gov/books/NBK1183/.
8. Robin NH, Moran RT, Ala-Kokko L. Sticklerin syndrooma. Jun. 9, 2000. . Julkaisussa: Pagon RA, Adam MP, Ardinger HH, et al., muokkaus. Yleisnäkemyksiä . Seattle (WA): University of Washington, Seattle; 1993-2014. Saatavilla: http://www.ncbi.nlm.nih.gov/books/NBK1302/.
9. Katsanis SH, Jabs YW. Treacher Collinsin Syndrooma. Heinäkuuta. 20, 2004. . Julkaisussa: Pagon RA, Adam MP, Ardinger HH, et al., muokkaus. Yleisnäkemyksiä . Seattle (WA): Washingtonin yliopisto, Seattle; 1993-2014. Saatavilla: http://www.ncbi.nlm.nih.gov/books/NBK1532/.
10. Schutte BC, Saal HM, Goudy s, et al. IRF6: een liittyvät häiriöt. MMA. 30, 2003. . Julkaisussa: Pagon RA, Adam MP, Ardinger HH, et al., muokkaus. Yleisnäkemyksiä . Seattle (WA): University of Washington, Seattle; 1993-2014. Saatavilla: http://www.ncbi.nlm.nih.gov/books/NBK1407/.