Mukopolysaccharidoses: early diagnostic signs in infants and children
Mukopolysaccharidoses (MPS) on ryhmä kliinisesti heterogeenisiä sairauksia, jotka johtuvat glykosaminoglykaanien hajoamiseen tarvittavien lysosomaalisten entsyymien puutteista.
MPS: lle on ominaista progressiiviset ja systeemiset kliiniset oireet. Biokemiallisesta ja geneettisestä heterogeenisuudestaan huolimatta eri tyyppien Keskeiset kliiniset piirteet vaihtelevat eri yhdistelmissä, mukaan lukien nivel-ja luustovika, johon liittyy jäykkyys (paitsi MPS IV, jossa on leväperäisyyttä) ja kipu, karkeat kasvonpiirteet, sarveiskalvon samentuminen, nivus-tai vatsatyrät, toistuvat ylähengitystieinfektiot, sydänläpän sairaus, rannekanavaoireyhtymä ja vaihteleva neurologinen osallistuminen. Nämä piirteet ilmenevät yleensä ensimmäisten elinkuukausien aikana vaikeiden muotojen osalta ja varhaislapsuudessa heikennetyimpien muotojen osalta, mutta ne usein aliarvioidaan ja otetaan huomioon yleensä vain silloin, kun ne ovat selvästi ilmeisiä.
näiden häiriöiden harvinaisuus ja kliinisen esitystavan vaihtelu johtavat usein diagnostiseen viiveeseen; vaikeat muodot voivat vaihdella kuukausista heikennettyihin, jopa vuosiin (KS. tässä lisäosassa). Kuitenkin, koska nopea eteneminen ja kiireellistä puuttumista vakavia esityksiä, jopa kuukausien viiveellä diagnoosi voi tuottaa katastrofaalisia tuloksia tulevan terveyden potilaan.
tavoitteenamme on korostaa tässä katsauksessa niitä hyvin varhaisia vaikeiden muotojen merkkejä, joiden pitäisi varoittaa lastenlääkäriä heidän ensiesiintymisestään.
- mitä merkkejä / oireita on odotettavissa lapsilla, joilla on vaikea MPS-muoto?
- mitkä ovat erityyppisten kansanedustajien erilaiset esitykset?
- MPS, joilla somaattinen ja kognitiivinen osallistuminen
- MPS, jolla oli lähinnä neurologisia ja kognitiivisia vaikutuksia
- MPS, joilla on vallitseva somaattinen vaikutus
mitä merkkejä / oireita on odotettavissa lapsilla, joilla on vaikea MPS-muoto?
MPS: n eri muotoihin viittaavat oireet ja merkit on esitetty taulukossa 1 iän mukaan. Ensimmäisen 6 kuukauden elämän, nivustyrät, poikkeuksellisen usein hengitystieinfektiot, otitis, ja organomegalia voidaan nähdä MPS potilailla, erityisesti MPS I (Hurler oireyhtymä), joka on kaikkein varhaiskypsä vaikea alkaa. Lisäksi 6-12 kuukauden kuluttua nämä lapset kehittävät hyvin usein gibbuksen (toraco-lannerangan kyfoosi), sydämen sivuäänen, napatyrän, lievän hypotonian ja kasvun viivästymisen . Niillä voi olla myös tyypillinen karkea kasvojen ulkonäkö(kuva. 1). MPS II, VI ja VII voivat esiintyä samanlaisella varhaisella fenotyypillä. MPS IV-potilailla on varhainen luustofenotyyppi, jossa lonkkavika ilmaantuu ensimmäisinä elinkuukausina ja kyyhkynrinta (pectus carinatum) ensimmäisten 12-18 kuukauden aikana. MPS II: n lapset ovat läsnä samalla tavalla osallistuneina, mutta myöhemmin ilmaantuvina. Toisen elinvuoden aikana MPS I Hurler syndrooma-vauvat kehittävät kognitiivista viivettä, kun taas MPS II-potilas voidaan tunnistaa vain umpeenkasvun, usein hengitystieinfektioiden ja useiden leikkausten vuoksi ; tyypilliset kasvonpiirteet näkyvät vasta myöhemmin. Joillakin MPS II-potilailla esiintyy myös hyperaktiivisuutta 18-24 kuukauden välillä. Kolmantena elinvuotena yliaktiivisuus ja kognitiivinen viive ovat helposti tunnistettavissa MPS II: ssa ja MPS III: ssa.
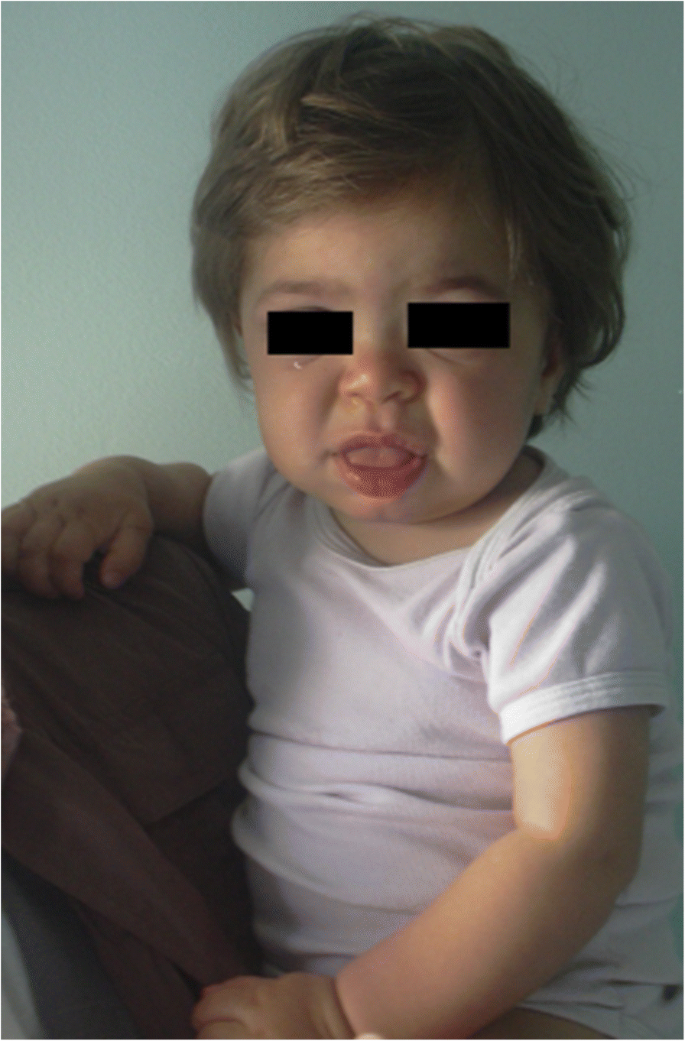
MPS IH vuonna 1-vuotias potilas. Karkeat piirteet: litteä nenäsilta, makroglossia, edestä pomottaminen
yhteenvetona voidaan todeta, että luuston ilmenemismuodot ovat tyypillisiä ja joskus varhaiskypsiä. Karkeat kasvot ja kognitiivinen viive eivät ole varhaisia näkyviä merkkejä.
mitkä ovat erityyppisten kansanedustajien erilaiset esitykset?
MPS, joilla somaattinen ja kognitiivinen osallistuminen
mukopolysakkaridoosi tyyppi I (MPS I; OMIM #252800) (viikunat. 1, 2, 3, 4 ja 5) aiheuttaa lysosomaalisen hydrolaasin α-l-iduronidaasin puutos, joka johtaa kahden Gag: n monisysteemiseen kertymiseen: dermataanisulfaatti (DS) ja heparaanisulfaatti (HS) . Vakavuuden perusteella MPS I on perinteisesti jaettu kolmeen kliiniseen fenotyyppiin; nämä muodot edustavat kuitenkin taudin vakavuuden jatkumoa. Hurlerin oireyhtymä, vakavin muoto, esiintyy tyypillisesti ensimmäisen elinvuoden aikana. Sairastuneet lapset kehittävät nopeasti merkittävää kognitiivista heikkenemistä ja somaattista sairautta useissa elinjärjestelmissä, mikä johtaa kuolemaan ensimmäisen vuosikymmenen aikana hoidon puuttuessa. MPS I: n heikennetyille muodoille, jotka tunnetaan Hurler–Scheien oireyhtymänä ja Scheien oireyhtymänä, on ominaista myöhempi oireiden alkaminen, pidempi elinajanodote ja lievä tai ei lainkaan keskushermoston (CNS) osallistuminen . Periytyvyys on autosomaalisesti resessiivinen, ja vähintään 50%: ssa tapauksista fenotyypin ennustaminen on mahdollista genotyypin perusteella, kun taas lopuissa 50%: ssa havaitaan uusia mutaatioita . Esiintyvyys on noin 1/100 000 elävänä syntynyttä .

MPS IH. Gibbus 9 kuukauden ikäisellä potilaalla. b selkärangan röntgenkuva 3-vuotiaan potilaan selkärangasta. c T2-painotettu magneettikuvaus selkärangasta, jossa on merkittävä kyfoosi ja nikamakanavan ahtauma

MPS IH. X-ray todisteita dysostosis multiplex. kylkiluiden paksuuntuminen 3-vuotiaalla potilaalla ja B lonkkavika toisella 18 kuukauden ikäisellä potilaalla

MPS IH: Kynsikäsi 18 kuukauden ikäisellä potilaalla; B käsiröntgen.

MPS IH. kallon kortikaalisen luun paksuuntuminen ja sella turcican poikkeava” J-muotoinen ” konformaatio. Periventrikulaaristen tilojen laajeneminen 17 kuukauden ikäisen potilaan B-ja C-T-2-painotetuissa magneettikuvauksissa
mukopolysakkaridoosi tyyppi II (MPS II; OMIM #309900) (Kuva. 6), joka tunnetaan myös Hunterin oireyhtymänä, johtuu iduronaatti-2-sulfataasientsyymin (I2S) puutteesta, josta seuraa DS: n ja HS: n GAG-kertymistä . Esiintyvyys on noin yksi 140 000-156 000 elävänä syntyneestä miehestä . Erityisesti, tämä on ainoa X-linkitetty MPS, ja naaras kantajat ovat yleensä oireettomia; kuitenkin, ne voivat poikkeuksellisesti vaikuttaa johtuen poikkeavuuksia X-kromosomi, homotsygositeetti, tai vinossa-X-inaktivaatio . Fenotyypin ilmentyminen kattaa myös laajan kirjon kliinistä vaikeusastetta. Vaikeissa tapauksissa esiintyy selvästi eteneviä neurologisia ja somaattisia sairauksia, ja kuolema tapahtuu yleensä elämän toisella vuosikymmenellä. Muilla potilailla, joilla on vain vähän tai ei lainkaan neurologisia toimintahäiriöitä, voi esiintyä vakavia somaattisia vaikutuksia, kun taas spektrin vastakkaisessa päässä on potilaita, joilla on vain minimaaliset somaattiset ilmentymät ja normaali älykkyys, jotka selviävät aikuisikään asti . Varhaiset merkit ja oireet jakavat MPS I: n ja II: n vaikeat muodot.

MPS II. 3-vuotiaalla potilaalla vain lieviä kasvonpiirteitä
MPS I: ssä kuvataan satunnaisia hydrops fetalis-tapauksia, vaikka yleensä nämä lapset vaikuttavat normaaleilta syntyessään. MPS I: n ensimmäiset merkit näkyvät yleensä aivan ensimmäisten elinkuukausien aikana, kun taas MPS II: n merkit ilmenevät yleensä hieman myöhemmin . Kiely ym. , retrospektiivisessä tutkimuksessa 55 hurler-potilaalla havaittiin, että hengitystieoireet, ruokintavaikeudet, nivustyrä ja välikorvatulehdus olivat ilmaantuneet mediaani-iässä ≤ 6 kuukautta, ja kyfoosi, sydämen poikkeavuudet, pään ympärysmitta, hypotonia, organomegalia, sarveiskalvon samentuminen, nivelrajoitus ja napatyrä ilmaantuivat 6 ja 12 kuukauden välillä. Napatyrä on yksi varhaisimmista MPS I: ssä ja II: ssa esiintyvistä piirteistä, ja nivustyriä raportoidaan noin 60%: lla potilaista .
toinen yleinen johtolanka MPS I: llä ja II: lla voi olla ylähengitysteiden toistuvat infektiot, joihin liittyy eritteiden lisääntymistä. Usein välikorvatulehdus, krooninen toistuva nuha ja jatkuva nenän vuotaminen ilman ilmeistä infektiota eivät ole spesifisiä, mutta voivat auttaa vahvistamaan diagnostista epäilyä, jos siihen liittyy tarkempia merkkejä. Gagin säilyttäminen Oro-nielussa johtaa risojen ja kitarisojen laajentumiseen ja edistää ylähengitysteiden komplikaatioita. Toinen usein esiintyvä havainto on meluisa hengitys, erityisesti yöllä, joka liittyy myöhemmissä vaiheissa obstruktiiviseen uniapneaan. Seurauksena usein välikorvan infektioita ja dysostosis välikorvan kuuloputket, kuulon heikkeneminen on usein, sekä johtava ja sensorineuraalinen alijäämät . Trakeobronchomalacia on yleisesti havaittu ja voi johtaa akuuttiin hengitysteiden ahtauma tai romahtaa, ja tämä on yksi tärkeimmistä kuolinsyistä seuraavina vuosina .
Adenoidektomia, nielurisaleikkaus ja tympanostomia sekä tyrän korjaus ovat MPS I-ja II-potilaille yleisempiä ja varhaisempia kirurgisia toimenpiteitä, jotka tehdään usein ennen diagnoosin saamista (yli 50%: ssa tapauksista) .
toistuva ripuli on melko yleistä MPS I: n alkuvaiheessa, ja sitä voi esiintyä myös MPS II-potilailla, joilla on neurologisia oireita, kun taas lievästi sairailla potilailla se on melko harvinaista .
Gibbuksen epämuodostuma (dorso-lannerangan kyfoosi) (Kuva. 2)ilmenee kliinisesti 8-vuotiaana.6 kk-1 vuosi MPS I: ssä, mikä edustaa varsin erityistä ja varhaiskypsää merkkiainetta kansanedustajista yleensä. Nikamarungot näyttävät litistyneiltä ja nokallisilta, mikä saattaa johtaa myöhempiin komplikaatioihin, kuten selkäydinhermon kiinnijäämiseen, akuuttiin selkäydinvammaan ja Atlanto-takaraivon epävakauteen; näin ollen on huolehdittava erityisesti atlanto-aksiaalinivelen sijoiltaanmenon estämisestä yleisanestesian ja leikkauksen aikana. 1 vuoden iän jälkeen nivelen kontraktuurat ja genu valgum ovat muita yleisiä merkkejä nivelen vajaatoiminnasta ja etenevästä luuston dysplasiasta, jotka ovat helposti havaittavissa alle 2 vuoden iässä kaikilla MPS: n sairastamilla lapsilla. Kaikki nämä luuston ilmentymät, jotka ovat tyypillisiä MPS: lle, ovat nimeltään dysostosis multiplex. Kylkiluiden paksuuntuminen näkyy röntgenkuvissa (Kuva. 3a), pitkät luut ovat lyhyitä leveine akseleineen, ja polvet ovat alttiita valguksen ja Varuksen epämuodostumille. Falangeaalinen dysostoosi ja synoviaalinen paksuuntuminen johtavat tyypilliseen kynnen käden epämuodostumaan (Kuva. 4), Mikä on toinen tyypillinen piirre, joka voi herättää epäilyksiä taudista. Lonkkavika on usein syy, miksi nämä lapset voidaan nähdä ensin ortopediset asiantuntijat. Tyypillisesti lantio on huonosti muodostunut, reisiluun päät ovat pienet ja coxa valga on yleinen, jossa on etenevä ja heikentävä lonkan epämuodostuma (Kuva. 3b) .
MPS I: n ja MPS II: n välillä on huomattava ero; MPS I: n luuston kasvu hidastuu 3 ikävuoteen mennessä, kun taas MPS II: n potilaiden liikakasvu ilmenee ensimmäisten 5-6 ikävuoden aikana , minkä jälkeen kasvu hidastuu .
kasvonpiirteiden (leveä nenä, jossa on levenevät sieraimet, huomattavat supraorbitaaliset harjanteet, suuret pyöristyneet posket, paksut huulet, laajentunut ulkoneva kieli) karkeus , joka johtuu Gagien varastoitumisesta orofasiaalisen alueen pehmytkudoksiin ja kasvojen luihin, ilmenee ensimmäisten 2 vuoden aikana, hurlerin taudin mediaaniajalla 1, 1 vuoden iässä ja Hunterin taudin vakavassa muodossa 18 kuukauden ja 4 vuoden välillä, joskin hienoisia fenotyyppisiä muutoksia voidaan havaita aikaisemmin (jo ensimmäisen vuoden jälkipuoliskolla) erityistä kokemusta omaavat lastenlääkärit (kuva. 1). Makrokefalia tulee vähitellen ilmeisemmäksi, ja scafosefalia on yleinen (kuva. 5), mutta mikrokefalia on mahdollista myös Hurler-potilailla . Kasvojen ja vartalon hirsutismi ilmenee aina 24 kuukauden ikään mennessä . Iho voi olla paksuuntunut ja joustamaton. Erityisesti, jotkut MPS II potilaat kehittävät erottuva iholeesio, joka on kuvattu norsunluunvalkoinen näppylöitä (Kivi-like) yläselässä ja sivuilla olkavarsien, patognomonic Hunter oireyhtymä .
kardiomyopatia ja etenevä venttiililehtisten paksuuntuminen ja jäykistyminen MPS I-potilailla voivat johtaa mitraaliin ja harvemmin aortan regurgitaatioon . Sivuääni voidaan havaita ensimmäisten elinkuukausien aikana, ja se on joskus raportoitu ensimmäisenä merkkinä taudista . Sydänoireita esiintyy myös lähes kaikilla MPS II-potilailla, mutta tämä alkaa noin 5-vuotiaana . Läppäsairaus voi johtaa oikean ja vasemman kammion hypertrofiaan ja sydämen vajaatoimintaan.
etenevän hepatosplenomegalian aiheuttama vatsan Protuberanssi on helposti havaittavissa 6 kuukauden iän jälkeen, joskaan Gag: iden säilyminen maksassa ja pernassa ei johda elinten toimintahäiriöihin .
sarveiskalvon samentumista esiintyy lähes kaikilla henkilöillä, joilla on MPS I alle 15 kuukauden iässä, mikä saattaa aiheuttaa vaikeaa näköhäiriötä. Päinvastoin, sarveiskalvon samentuminen ei ole tyypillinen piirre Hunterin oireyhtymässä , mutta rakolampun tutkimus voi paljastaa erillisiä sarveiskalvon vaurioita, jotka eivät vaikuta näkökykyyn. Verkkokalvon toimintahäiriö voidaan havaita, kun taas glaukooma ei ole yleinen löydös.
varhainen psykomotorinen kehitys havaitaan yleensä normaaliksi, mutta MPS I: llä kehitysviive on yleensä ilmeinen 18 kuukauden ikään mennessä, ja asteittainen henkinen lasku johtaa vakavaan kehitysvammaisuuteen. Kielitaito on näillä lapsilla hyvin rajallinen. MPS II: n kehityksen virstanpylväiden maailmanlaajuinen viivästyminen ilmenee 2 vuodesta eteenpäin . Vakavan Hunterin taudin johtavaa neurologista piirrettä edustavat kuitenkin huomattavat käytöshäiriöt, kuten yliaktiivisuus, itsepäisyys ja aggressiivisuus, joita ei tyypillisesti havaita niillä, joilla on heikennetty fenotyyppi .
mukopolysakkaridoosi tyyppi VII (MPS VII; OMIM #253220), joka tunnetaan myös nimellä Sly-oireyhtymä, on erittäin harvinainen MPS, jolle on ominaista β-glukuronidaasin puutteellinen aktiivisuus, kondroitiinisulfaatin (CS), DS: n ja HS: n lysosomaalinen varastointi, mikä johtaa solujen ja elinten toimintahäiriöihin. Ensimmäisen potilaan kuvasi Sly et al. vuonna 1973 ja tähän mennessä kirjallisuudessa on raportoitu yhteensä 145 potilaasta.
MPS VII: n kliininen esiintymistapa ja taudin eteneminen ulottuu laajaan vaikeusasteeseen varhaisista, vaikeista monijärjestelmäoireista ja ensimmäisten kuukausien kuolemista lievempään fenotyyppiin, joka puhkeaa myöhemmin, jonka älykkyys on normaali tai lähes normaali ja jossa eloonjääminen on pitempää .
MPS VII-potilaiden fenotyyppiset ominaisuudet muistuttavat MPS I: n ja MPS II: n fenotyyppejä (lyhytkasvuisuus, luuston dysplasia, makrokefalia, ientulehdusten hypertrofia, toistuvat korvatulehdukset, hepatosplenomegalia, tyrät, sydänoireet, heikentynyt keuhkojen toiminta ja kognitiivisten kykyjen heikkeneminen). Odottamattoman suurella osalla kuvatuista potilaista (41%) on ollut ei-immuuni vastasyntyneen hydrops fetalis (nihf) . Vaikka tauti puhkesi näillä potilailla ennen syntymää, 13 potilasta 23: sta selviytyi vauvaiästä lievällä tai keskivaikealla tavalla myöhäiseen teini-ikään. Näin ollen nihf: n läsnäolo ei itsessään ennusta kliinisen kurssin mahdollista vakavuutta, jos potilas selviää vauvaiässä. Toinen varhainen merkki on makrokrania, kun taas sydänläpän osallistuminen ja toistuvat infektiot ylä-ja alahengitysteiden näkyvät ensimmäisen 2 elinvuoden aikana. Myös keskivaikea kehitysvammaisuus ja etenevä kuulonalenema puhevammoineen näkyvät ajan myötä.
yhteenvetona voidaan todeta, että MPS I: n vaikeat muodot puhkeavat hyvin varhaisessa vaiheessa (6 kuukauden sisällä elämästä); MPS II on samanlainen, mutta myöhemmin alkava, ja MPS VII: llä on prenataalinen alkaminen, jossa on usein NIHF: ää ja varhainen vakava ylä-ja alahengitysteiden osallistuminen.
MPS, jolla oli lähinnä neurologisia ja kognitiivisia vaikutuksia
Mukopolysakkaridoosit tyyppi III (MPS III, tunnetaan myös nimellä Sanfilippon oireyhtymä; viikuna. 7) on vallitseva neurologinen esitys. MPS III on lysosomaalinen säilytyshäiriö, joka johtuu yhden HS: n kataboliaan osallistuvan neljän entsyymin puutoksesta. Neljä eri alatyyppiä tunnetaan—MPS III tyyppi A (OMIM #252900), tyyppi B (OMIM #252920), tyyppi C (OMIM #252930) ja tyyppi D (OMIM #252940)—kukin johtuu eri entsyymin puutoksesta: Tyyppi a, sulfamidaasi/sgsh-geeni; tyyppi B, alfa-N-asetyyliglukosaminidaasi/NAGLU-geeni; tyyppi C, alfa-glukosaminidi N-asetyylitransferaasi/HGSNAT geeni; tyyppi D, n-ACETILGLUCOSAMINA-6-solfato sulfatasi/GNS geeni). Kaikilla neljällä tyypillä (A-D) on autosomaalinen resessiivinen periytyminen. Kansanedustajista yleisin on MPS III, jonka esiintyvyys on arviolta 0,3-4.1 tapaus jokaista 100 000 vastasyntynyttä kohti, riippuen alatyypistä ja rodusta .

MPS IIIA. Sama potilas 2,5-ja b 5-vuotiaana. Huomaa erilainen kasvojen ilme, joka osoittaa neurologisen vajaatoiminnan etenemistä
Sanfilippon oireyhtymää sairastavilla potilailla ilmenee etenevää kognitiivista heikentymistä toisen ja kolmannen elinvuoden aikana. Toisin kuin valtaosalla kansanedustajista somaattiset piirteet ovat suhteellisesti vähemmän havaittavissa, ja KESKUSHERMOSTOVAIKUTUS on silmiinpistävää. Puheen kehityksen viivästyminen on yleensä ensimmäinen merkki, jonka vanhemmat huomaavat, mutta tärkeimmät huolenaiheet voivat olla käyttäytymisongelmat (hyperaktiivisuus, ahdistunut ja aggressiivinen käytös, unihäiriöt), jota seuraa progressiivinen henkinen heikkeneminen . Nämä oireet voivat aiheuttaa vääriä diagnooseja käytöshäiriöinä, tarkkaavaisuus-ja ylivilkkaushäiriöinä (ADHD) ja autismin kirjon häiriöinä . Myös epilepsiaa voi esiintyä.
unihäiriöt , joiden esiintyvyys on raportoitu jopa 80-90%, ovat yleinen piirre. Ne koostuvat nukahtamisvaikeuksista ja toistuvasta yöllisestä heräämisestä, ja joillakin potilailla päivä–yö-rytmi on täysin toisinpäin.
kaikkia näitä ilmenemismuotoja on hyvin vaikea hallita, ja niillä on valtava psykologinen vaikutus koko perheen elämänlaatuun.
yleisesti ottaen, vaikka fenotyyppi voi olla hyvin samankaltainen neljällä alatyypillä, Sanfilippo B: n ja C: n kliiniselle kurssille näyttää olevan ominaista lievempi fenotyyppi .
ei-neurologiset oireet ovat yleensä lievempiä kansanedustajilla III kuin muilla kansanedustajilla. Toistuvia Korva -, nenä-ja kurkkutulehduksia havaitaan nuoremmalla iällä. Toistuvat korvatulehdukset, välikorvan kuulovammat ja sisäkorvan poikkeavuudet voivat aiheuttaa kuuroutta. Ylimääräinen paksu eritteiden ja anatomiset muutokset voivat aiheuttaa tukkeutumisen hengitysteiden. Ensimmäisinä elinvuosina ripulia esiintyy usein, kun taas ummetus on yleisempää vanhemmilla potilailla.
lääkärintarkastuksessa voi näkyä karkeita kasvonpiirteitä, joskin ne ovat lievempiä. Potilailla on makrokefalia, leveät kulmakarvat mediaalineulontoineen ja synofrys, hiukset ovat yleensä kuivat ja karkeat, ja hypertrichosis on yleistä. Nuorilla potilailla havaitaan usein lievää hepatomegaliaa ja napanuora-ja nivustyriä. Harvinaisia ja usein lieviä kontraktuuroja on lähinnä kyynärpäissä. Kasvua esiintyy noin puolella yli 12-vuotiaista potilaista .
yhteenvetona voidaan todeta, että MPS III: lla tai Sanfilippon taudilla on huomattava neurologinen vaikutus, johon liittyy hyvin lieviä somaattisia oireita. MPS III: lla voidaan epäillä olevan 2-3-vuotiaita pikkulapsia, joilla esiintyy hyperaktiivisuutta, ADHD: tä ja aggressiivista käyttäytymistä.
MPS, joilla on vallitseva somaattinen vaikutus
MPS IV ja MPS VI, joilla on vain somaattinen vaikutus. Nämä ovat asteittaisia tiloja, jotka säästävät älyllisiä kykyjä ja vaikuttavat pääasiassa luurankoon (MPS IVA) tai kaikkiin kehon elimiin/järjestelmiin (MPS VI). Periytyvyys on autosomaalisesti resessiivinen. Nopeus, jolla oireet pahenevat vaihtelee vaikuttaa yksilöiden.
mukopolysakkaridoosi IV (MPS IV, tunnetaan myös nimellä Morquion oireyhtymä; viikuna. 8) esiintyy kahtena geneettisesti erillisenä häiriönä, joilla molemmilla on erilainen entsyymin puutos: MPS IVA (Morquio a-oireyhtymä; OMIM #253000), joka johtuu n-asetyyligalaktosamiini-6-sulfataasin (GALNS-geeni) puutteesta, mikä johtaa kerataanisulfaatin (KS) ja CS ; ja MPS IVB (Morquio B-oireyhtymä; OMIM #253010), joka johtuu beeta-galaktosidaasiaktiivisuuden puutteesta, joka johtaa virtsaan KS ja oligosakkaridin erittymiseen kuten GM1-potilailla. MPS IVB On todellakin alleeli GM1-gangliosidoosin eri muodoille, mutta siitä puuttuu GM1-gangliosidoosissa havaittu psykomotorinen heikkeneminen .

MPS IVA. 4-vuotias potilas, jolla B-antero-posteriorinen ja c-lateraalinen röntgenkuva, jossa näkyvät “melan muotoiset” kylkiluut ja litistetyt ja pyöristetyt nikamat, joilla on tyypillinen “etumainen nokan muodostus” – näkökulma.
potilaat, joilla on vaikea MPS IVA ja joilla ei ole havaittavaa entsyymiaktiivisuutta, paljastavat ensimmäiset oireensa yleensä ensimmäisen elinvuoden aikana, vaikka ne diagnosoidaan harvoin ennen 4-5 vuoden ikää. Tiedot morquio A-potilaiden luonnonhistoriasta ovat peräisin pääasiassa kahdesta laajasta aineistokokoelmasta . Kasvun hidastuminen alkaa noin 18 kuukauden iässä ja kasvu pysähtyy noin 7 tai 8 vuoden iässä. Toisin kuin muut MPS, Morquio a nivelet ovat yleensä löyhiä ja hyvin joustava (hypermobile), mutta joissakin nivelissä (yleensä lantion ja hartioiden) voi olla rajoitettu liikerata. Potilailla, joilla on tällainen luustoon liittyvä sairaus, voidaan diagnosoida spondyloepifyseaalinen dysplasia, pseudoachondroplasia, multippeli epifyseaalinen dysplasia tai bilateraalinen Legg-Calvé-Perthes-tauti tai heille voidaan tehdä sen arviointi .
jatkuva piirre on odontoidinen hypoplasia; näin ollen Atlantiksen ja niskanivelen sijoiltaanmeno voi aiheuttaa puristusta ja vaurioita bulbaariin ja selkäytimeen, mikä johtaa halvaantumiseen tai jopa kuolemaan, jos kohdunkaulan vakauttamista ei tehdä aikaisin.
MPS IVB-potilailla on hyvin samanlainen kliininen ilmentymä, mutta vähemmän näkyvä lyhytkasvuisuus. Heillä on lievästi karkeat kasvonpiirteet, kuulon heikkeneminen, sarveiskalvon samentumat, läppävika ja usein esiintyviä ylähengitystieinfektioita. Ne voivat myös esiintyä nivustyrä ja lievä hepatomegalia. Luuston dysplastisiin ominaisuuksiin kuuluvat platyspondylia, odontoidinen hypoplasia, johon liittyy mahdollisesti kohdunkaulan subluksaatio, kypho-skolioosi, coxa valga ja ahtaat suoliluun Siivet .
mukopolysakkaridoosi VI (MPS VI, tunnetaan myös nimellä Maroteaux-Lamyn oireyhtymä; OMIM #253200; Fig. 9) johtuu kromosomissa 5 sijaitsevan aryylisulfataasi B: n (ARSB) geenin patogeenisistä mutaatioista. Tämän seurauksena aryylisulfataasi B (ASB)-entsyymin (N-asetyyligalaktosamiini-4-sulfataasi) vähentynyt tai puuttuva aktiivisuus heikentää DS: n hajoamista määrittämällä sen solujen kertymistä .

MPS VI. 2-vuotias poika ilmeinen karkea kasvot ja luuston dysostosis
potilailla, joilla ei ole havaittavaa entsyymiaktiivisuutta, on vaikea MPS VI-muoto ja oireita varhaislapsuudessa. Niiden somaattinen fenotyyppi muistuttaa hurlerin oireyhtymää. Useimmissa tapauksissa, 2 tai 3 vuoden iässä, vakava ja etenevä luustovaurio, nimeltään dysostosis multiplex, ilmenee. Tämä luuston dysplasia sisältää lonkkavika dysplastinen reisiluun pää, epänormaali selkärangan elinten, epäsäännölliset solisluut, hypoplastinen distaalinen kyynärluu ja säde, ja dysplastinen ja lyhyt metakarpaalinen luut; ranneke ja tarsal luut ovat hypoplastinen ja on epäsäännöllinen profiili. Kasvunopeus hidastuu usein ensimmäisen elinvuoden jälkeen, ja lopullinen pituus on yleensä alle 120 cm. Kuten muillakin kansanedustajilla, varhainen merkki ovat karkeat kasvonpiirteet. Lisäksi potilailla on tyypillinen ulkoneva vatsa, hepatomegalia, napanuora ja/tai nivustyrä ja sydämen osallistuminen varhaislapsuudessa. Vaikka primaarista kehitysvammaisuutta ei esiinny, joillakin potilailla voi esiintyä hydrokefalusta, josta seuraa kallonsisäinen hypertensio ja papilledeema. Pectus carinatum, jäykät ja supistuneet nivelet, skolioosi ja kyfoosi sekä kohdunkaulan selkäytimen puristus pahenevat ajan myötä .
yhteenvetona voidaan todeta, että kansanedustajilla IV ja VI ei ole keskushermostoon liittyviä vaikutuksia. MPS IV on ainutlaatuinen MPS osoittaa yhteinen löystyminen, kun taas vakava MPS VI puhkeaminen on samanlainen kuin havaittu MPS I.