Frontiers in Neurology
Einleitung
Neuroacanthocytosis ist ein übergeordneter Begriff für eine Gruppe seltener Syndrome, die durch neurologische Symptome in Kombination mit stachelig deformierten roten Blutkörperchen (Akanthozyten) gekennzeichnet sind. Dieser Bericht konzentriert sich auf die Chorea-Akanthozytose (ChAc) , eine seltene Erkrankung mit schätzungsweise 1.000 betroffenen weltweit, die durch autosomal-rezessive Mutationen im VPS13A-Gen (Vacuolar Protein Sorting 13 Homolog A) auf Chromosom 9q (1) verursacht wird. Die Krankheit verläuft progressiv, Ursachen für eine erhöhte Mortalität können verminderte motorische Funktionen sein, die mit Risikozuständen wie Dysphagie korrelieren, aber auch plötzliche unerwartete Todesfälle wurden beschrieben (2, 3). Bisher gibt es keine ursächliche Behandlung.
Die verdächtige Koexistenz von Akanthozytose und Bewegungsstörungen wurde erstmals in den 1970er Jahren von Levine und Critchley berichtet (4, 5) und wurde seitdem als herausragendes Merkmal der Krankheit akzeptiert. Mit unserem Fallbericht beschreiben wir jedoch einen seltenen klinischen Phänotyp von ChAc ohne offensichtliche Bewegungsstörungen, was auf eine breitere Vielfalt klinischer Präsentationen hindeutet. Diagnostische Werkzeuge müssen sich diesen Herausforderungen stellen, um die richtige Diagnose zu stellen, Wir schlagen hier molekulare Neuroimaging als Schlüsselmethode vor.
Fallbericht
Der männliche Indexpatient zeigte erstmals im Alter von 25 Jahren zwei unprovozierte bilaterale tonisch-klonische Anfälle. Es gab keine Krankengeschichte. Der MRT-Scan des Gehirns und das EEG in diesem Alter waren normal und aufgrund der Zurückhaltung des Patienten und seltener Ereignisse wurde keine Behandlung eingeleitet. Der Patient hatte jedoch anhaltende Anfälle, die sich jetzt als partielle Epilepsie darstellten. Er beschrieb wiederkehrende Gefühle von plötzlichem Schwindel, die als schwindelerregende Aura diagnostiziert wurden. Darüber hinaus hatte er dyskognitive Anfälle, bei denen er entweder (a) nicht reagierte und türkische Gebete rezitierte, (b) mündliche Automatismen oder (c) mit den Händen hantierte und Geräusche aussprach — alles teilweise in tonisch-klonische Anfälle. Das Video-EEG zeigte nun lokale Spike-Wave-Komplexe sowohl im rechten als auch im linken Temporallappen. Entsprechend der Semiologie des Anfalls wurde die Diagnose einer bilateralen Temporallappenepilepsie gestellt und mit der Medikation mit Levetiracetam begonnen.
Im Alter von 31 Jahren wurden die Anfälle nicht vollständig unterdrückt, im Rahmen der weiteren Diagnostik der Temporallappenepilepsie wurde der Patient einem FDG-PET unterzogen (Abbildung 1), das einen bilateralen mesiotemporalen Hypometabolismus zeigte, der dem mesiotemporalen Anfall entsprach Ursprung. Es gab jedoch auch einen ausgeprägten, sehr ungewöhnlichen striatalen Hypometabolismus, der den Verdacht auf eine neurodegenerative Bewegungsstörung aufkommen ließ. Daher wurde eine umfangreiche Follow-up-Evaluation eingeleitet (siehe Abbildung 2). Es wurde eine FP-CIT-SPECT durchgeführt, die einen mäßig bilateralen Verlust der Verfügbarkeit des striatalen Dopamintransporters im Einklang mit einer Abnahme der nigrostriatalen Integrität ergab (Abbildung 1). Eine hochauflösende MRT zeigte nun eine bilaterale Atrophie des Nucleus caudatus (Abbildung 3). Die neurologische Untersuchung zeigte ein diskretes gebundenes Gangmuster und Hypomimie, aber keine andere Erkrankung des extrapyramidalen motorischen Systems und keine bulbären Symptome wie Muskeldystonie. Abgesehen von einem niedrigen Reflexstatus an den unteren Extremitäten, bei dem Nervenleitungsstudien eine sensomotorische axonale Polyneuropathie bestätigten, war die neurologische Untersuchung normal. Zu den neuropsychologischen Symptomen gehörten Aspekte einer Angststörung, und der Patient berichtete über Schwierigkeiten mit Kurzzeitgedächtnisverlust. Neuropsychologische Untersuchungen bestätigten jedoch keine manifeste Gedächtnisfehlfunktion, sondern eine unspezifische Ablenkung, die möglicherweise zusätzlich durch eine unzureichende Anfallsunterdrückung verstärkt wurde. Labortests waren negativ für jede symptomatische Epilepsie (d.h., Autoimmun-Enzephalitis-Antikörper, antineuronale Antikörper). Liquor cerebrospinalis zeigte keine Anzeichen einer Entzündung, aber einen moderaten Proteinanstieg (765 mg / l). Die Analyse von biogenen Aminen in der Zerebrospinalflüssigkeit ergab eine Erhöhung von Glutamin, die wir mit den jüngsten Anfällen in Verbindung brachten. Im peripheren Blut wurden 0,4% Akanthozyten gefunden. Serologische Tests auf Morbus Wilson waren negativ. Auffällig war eine anhaltende Erhöhung der Kreatinkinase (Bereich: 1.000–3.000 U/l), die zur Verdachtsdiagnose einer mitochondrialen Störung führte. Für weitere Untersuchungen wurde ein ischämischer Laktattest durchgeführt, der normale Ergebnisse zeigte. Eine Muskelbiopsie des M. gastrocnemicus wurde durchgeführt, aber die Analyse der Mitochondrienfunktion und der Atmungskette war normal.
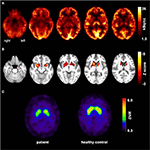
Abbildung 1. Ergebnisse von molekularen Bildgebungsstudien. Transaxiale FDG-PET-Bilder (A) zeigten nicht nur einen leichten bilateralen mesiotemporalen Hypometabolismus (im Einklang mit dem mesiotemporalen Anfall), sondern auch einen ausgeprägten striatalen Hypometabolismus, der im Vergleich zu gesunden Kontrollen als hoch signifikant befunden wurde . Eine zusätzliche FP-CIT-SPECT-Untersuchung (C) ergab auch einen moderaten Verlust von striatalen Dopamintransportern, was zu einer Abnahme der nigrostriatalen Integrität führte (zum Vergleich wird eine altersgerechte gesunde Kontrolle gezeigt; DVR, Verteilungsvolumenverhältnis).

Abbildung 2. Klinischer und diagnostischer Verlauf des Indexpatienten.
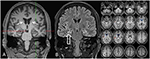
Abbildung 3. Die MRT zeigt eine diskrete bilaterale Schwanzkopfatrophie und einen kleineren und hyperintensiven rechtsseitigen Hippocampus, der auf eine rechtsseitige Hippocampus-Sklerose hinweist . Der linke Hippocampus ist etwas hyperintensiv, aber nicht atrophisch. Die bilaterale Schwanzkopfatrophie wird durch eine CVR-Analyse bestätigt, bei der blaue Farben ein vermindertes graues und rote Farben ein erhöhtes Liquorvolumen aufweisen (C).
Die Familiengeschichte enthüllte eine Blutsverwandtschaft seiner Eltern (Cousins ersten Grades), die selbst keine signifikante Krankengeschichte hatten. Von seinen sechs Geschwistern hatten zwei (im Alter zwischen 20 und 30 Jahren) inzwischen ebenfalls an allgemeinen Anfällen gelitten (siehe Abbildung 4). Krankenakten seiner Geschwister lagen uns nicht vor.
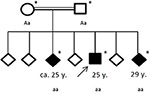
Abbildung 4. Stammbaum der betroffenen Familie, mit Alter des ersten epileptischen Anfalls in Jahren (y). Pfeil: Index Patent. Sternchen: Gentests durchgeführt. Aa, heterozygot; aa, homozygot für c.4326 T>A (s.11442*).
Unter der Hypothese einer erblichen neurodegenerativen Bewegungsstörung wurden der Patient und seine Familie zur genetischen Untersuchung überwiesen. Die Exomsequenzierung ergab eine homozygote, trunzierende Mutation c.4326 T>A (p.Tyr1442*) im ChAc-Gen VPS13A bei den drei betroffenen Geschwistern. Die blutsverwandten Eltern wurden beide heterozygot nachgewiesen. Bei der Exomsequenzierung wurden keine weiteren Varianten oder Mutationen identifiziert.
Tatsächlich hatte der Indexpatient in einer Nachuntersuchung im Alter von 33 Jahren immer noch nur marginale extrapyramidale Symptome und bei keiner seiner Schwestern waren diese vorhanden. Er erweckte nun den Eindruck einer leichten Starrheit nur unter Grundierung an der rechten oberen Extremität und einer leichten Bradykinesie der oberen Extremitäten. Unter der Therapie mit Levetiracetam und Lacosamid war der Patient anfallsfrei.
Diskussion
Mit dem vorliegenden Fall zeigen wir, dass sich ChAc klinisch bei Temporallappenepilepsie ohne signifikante motorische Symptome aufgrund einer neuen entsprechenden Mutation im VPS13A-Gen manifestieren kann.
Der Krankheitsverlauf ist typischerweise durch eine fortschreitende Bewegungsstörung (einschließlich Chorea, Dystonie, Parkinson), kognitive und psychiatrische Veränderungen und myopathische Symptome mit serologischen Markern für Akanthozytose und Hyperglykämie gekennzeichnet. Krampfanfälle wurden zuvor als Symptom von ChAc festgestellt (6), aber Bewegungsstörungen werden immer noch als Schlüsselsymptom angesehen. Es gibt Hinweise darauf, dass Epilepsie und insbesondere Epilepsie mit Ursprung im Temporallappen ein unterschätzter Phänotyp von ChAc sein könnte. Peluso et al. berichtete über einen Patienten ähnlich unserem, der selbst im Alter von 46 Jahren keine Bewegungsstörungen zeigte, während er Neuroimaging-Befunde vorlegte, die auf eine Beteiligung der Basalganglien hindeuteten (7). In Übereinstimmung damit haben Scheid et al. beschrieben drei Patienten, die an mesialer Temporallappenepilepsie und Sklerose litten (basierend auf MRT-Studien) als vorherrschendes Symptom und wurden mit ChAc diagnostiziert (8). Darüber hinaus haben Mente et al. kürzlich vorgelegte Autopsieergebnisse eines ChAc-Patienten, der nicht nur eine Basalganglienatrophie, sondern auch eine Hippocampus-Sklerose aufwies (9). In Übereinstimmung mit unserem Fall scheint die bilaterale Beteiligung des Hippocampus ein häufiger Befund zu sein (10, 11). Der genaue Zusammenhang zwischen Anfällen und Sklerose ist jedoch immer noch eine Frage von Henne oder Ei. Die korrelierende Rolle von Chorein in der strukturellen Entwicklung des Hippocampus ist noch nicht vollständig verstanden. Interessanterweise wurde in einem Mausmodell von ChAc eine strukturelle Veränderung beobachtet, da die Hippocampus-Proteinexpression des GABA (A) -Rezeptors gamma 2 und seines Ankerproteins Chorein erhöht waren (12).
In der klinischen Routine kann dieser Phänotyp immer noch eine Herausforderung bei der Suche nach der richtigen Diagnose darstellen. Der erste entscheidende Hinweis auf eine neurodegenerative Erkrankung bei unserem Patienten war ein deutlicher Rückgang des striatalen Glukosestoffwechsels und ein moderater Rückgang der nigrostriatalen Integrität bei FDG-PET bzw. Die kleine Sammlung von funktionellen Bildgebungsergebnissen, basierend auf Fallserienbeschreibung, zeigen, dass Veränderungen im Stoffwechsel und dopaminerge Dysfunktion treten meist im Nucleus caudatus und Putamen auf (13). Die MRT des Gehirns zeigte im Verlauf der Erkrankung eine Atrophie des Nucleus caudatus, die als typischer Befund bei ChAc beschrieben wurde (14). Dementsprechend umfassen unterschiedliche neurologische Merkmale normalerweise Chorea, Ernährungsdystonie, orofaciolinguale Dyskinesien und Tics sowie andere Symptome einer Basalganglienerkrankung wie Parkinsonismus und Dystonie (3). Es ist verlockend zu spekulieren, dass die beobachteten striatalen Veränderungen bei unserem Patienten immer noch unter der Schwelle lagen, Symptome zu verursachen (d. H. prodromale Bildgebungsbefunde), die letztendlich mit fortschreitender Krankheit auftreten können. Wir fördern daher die strukturelle und molekulare Bildgebung als sensitives diagnostisches Instrument bei dieser seltenen Erkrankung.
In ChAc gibt es Beschreibungen von Akanthozytenzahlen zwischen 5 und 50% (3), aber es gibt einige Fallberichte, die zeigen, dass Akanthozyten möglicherweise erst spät im Krankheitsverlauf auftreten (15), sehr niedrig sind oder sogar vollständig fehlen (16). Daher sollten Akanthozyten für die Diagnose nicht als obligatorisch angesehen werden. Darüber hinaus kann Epilepsie die Diagnose verzögern, da sie andere typische Symptome von ChAc verschleiert. Insbesondere psychiatrische Symptome wie Persönlichkeitsveränderungen und kognitive Verschlechterung, die sehr häufig auftreten, können falsch interpretiert und auf arzneimittelbedingte Nebenwirkungen (d. H. von Levetiracetam) oder anfallsbedingte Nebenwirkungen zurückgeführt werden. Sekundäre Hyperglykämie tritt nach allgemeinen tonisch-klonischen Anfällen auf, und eine anhaltende Erhöhung kann übersehen werden.
Unser Fall hebt auch den entscheidenden Vorteil der Exomsequenzierung hervor, um eine korrekte Diagnose zu stellen, insbesondere bei seltenen Krankheiten oder wenn die Symptome vage sind. Die Familie der Säugetier–VPS13 (A-D) -Gene ist von wachsendem Interesse, und Mutationen wurden bei anderen neurologischen Erkrankungen wie der autosomal-rezessiven Parkinson-Krankheit oder der autosomal-rezessiven spinocerebellären Ataxie identifiziert (17). Rezessive Mutationen in VPS13D verursachen Bewegungsstörungen im Kindesalter (18). Die zugrunde liegende Pathophysiologie von ChAc ist noch nicht vollständig entschlüsselt, aber es wurde gezeigt, dass VPS13A das Protein Chorein kodiert, das im Gehirn und in einer Vielzahl anderer Gewebe ubiquitär exprimiert wird (19). Studien zeigen, dass es an Signalwegen beteiligt ist, die die Architektur des Zytoskeletts, die Exozytose und das Überleben der Zellen regulieren (20). Eine weitere Mutation, die bei 9 Patienten mit ChAc mit einem anfallsdominierten Phänotyp beschrieben wurde, ist die c.2343del-Mutation im VPS13A-Gen (21). Es ist vernünftig anzunehmen, dass spezifische Mutationen im selben Gen zu einer bestimmten Aberration von Chorein führen, die einen einzigartigen Phänotyp verursacht, indem sie beispielsweise verschiedene Bereiche des Gehirns betreffen. Die zugrunde liegende Pathophysiologie ist jedoch nur spekulativ und eine weitere Dokumentation der ursächlichen Mutationen wird von Interesse sein. Wir zeigen hier, dass die bisher nicht beschriebene trunzierende Mutation c.4326 T>A (p.Tyr1442*) bei allen betroffenen Familienmitgliedern zu einem ähnlichen Phänotyp mit vorherrschender Epilepsie führt.
Abschließende Bemerkungen
Epilepsie (insbesondere bilaterale Temporallappenepilepsie) ist wahrscheinlich ein vorherrschender Phänotyp von ChAc. Daher sollte eine Suche nach zusätzlichen roten Fahnen (wie Hyperglykämie, Familienanamnese, Polyneuropathie) sorgfältig durchgeführt werden. In verdächtigen Fällen Suche nach Akanthozyten im Blut und Gehirn MRT sollte hinzugefügt werden, da diese Werkzeuge weit verbreitet sind. Im Zweifelsfall sollte die Diagnostik mit FDG-PET und / oder FP-CIT-SPECT ergänzt werden, da sie empfindlich auf eine striatale Beteiligung reagieren. Bei autosomal-rezessiven Epilepsien mit indikativem Befund für eine neurodegenerative Erkrankung sollte dann der VPS13A-Einzelgen-Test oder Chorein-Western-Blot verwendet werden, um die richtige Diagnose zu stellen. Letzteres sollte auch bei anderen neurodegenerativen Erkrankungen ohne genetisch definierte Ätiologie in Betracht gezogen werden.
Ethikerklärung
Für die Teilnahme an der Studie wurde vom Patienten eine schriftliche Einverständniserklärung eingeholt. Für die Veröffentlichung dieses Fallberichts wurde vom Patienten eine schriftliche Einverständniserklärung eingeholt.
Autorenbeiträge
JW, MR und SK nahmen am Patientenmanagement teil. JW schrieb den ersten Entwurf des Manuskripts. LF, HU und PM bereiteten Zahlen vor. Alle Autoren haben zur Überarbeitung des Manuskripts beigetragen, die eingereichte Version gelesen und genehmigt.
Interessenkonflikterklärung
Die HU ist Gesellschafterin der Veobrain Gmbh, einem Spin-off der Universitätsmedizin Freiburg.
Die übrigen Autoren erklären, dass die Forschung in Abwesenheit von kommerziellen oder finanziellen Beziehungen durchgeführt wurde, die als potenzieller Interessenkonflikt ausgelegt werden könnten.
1. Walterfang M, Evans A, Leong Looi JC, Jung HH, Danek A, Walker RH, et al. Die Neuropsychiatrie der Neuroakanthozytosesyndrome. Neurosci Biobehav Rev. (2011) 35:1275-83. doi: 10.1016/j.neubiorev.2011.01.001
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
2. Walker RH. Management von Neuroakanthozytosesyndromen. Tremor Andere Hyperkinet. Mov. (2015) 5:346. doi: 10.7916/D8W66K48
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
3. Es sind keine frei zugänglichen ergänzenden Materialien verfügbar in: Velayos Baeza A, Dobson-Stone C, Rampoldi L, Walker RH, Danek A, et al. Chorea-Akanthozytose. GeneReviews®. Seattle, WA: Universität von Washington (1993).
4. Critchley EM, Clark DB, Wikler A. Akanthozytose und neurologische Störung ohne Betalipoproteinämie. Arch Neurol. (1968) 18:134–40.
PubMed Zusammenfassung
5. Levine IM, Estes JW, Looney JM. Erbliche neurologische Erkrankung mit Akanthozytose. Ein neues Syndrom. Arch Neurol. (1968) 19:403–9.
PubMed Zusammenfassung / Google Scholar
6. Hardie RJ, Pullon HW, Harding AE, Owen JS, Pires M, Daniels GL, et al. Neuroakanthozytose. eine klinische, hämatologische und pathologische Untersuchung von 19 Fällen. Gehirn (1991) 114(Pt 1A): 13-49.
PubMed Zusammenfassung / Google Scholar
7. Esposito M, Antennora A, Pappata S, Rosa ADR, et al. Chorea-Akanthozytose ohne Chorea: Erweiterung des klinischen Phänotyps. In: Parkinson Relat Disord. (2017) 41:124–6. geburtsdatum: 10.2016/j.parkreldis.2017.05.013
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
8. Scheid R, Bader B, Ott DV, Merkenschlager A, Danek A. Entwicklung der mesialen Temporallappenepilepsie bei Chorea-Akanthozytose. Neurologie (2009) 73: 1419-22. Ursprungsbezeichnung: 10.1212/WNL.0b013e3181bd80d4
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
9. Mente K, Kim SA, Grunseich C, Hefti MM, Crary JF, Danek A, et al. Hippocampus-Sklerose und mesiale Temporallappen-Epilepsie bei Chorea-Akanthozytose: ein Fall mit klinischer, pathologischer und genetischer Bewertung. In: Neuropathol Appl Neurobiol. (2017) 43:542–6. doi: 10.1111/nan.12403
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
10. Bader B, Vollmar C, Ackl N, Ebert A, la Fougère C, Noachtar S, et al. Bilaterale Temporallappenepilepsie mit intrakraniellem EEG bei Chorea-Akanthozytose bestätigt. Beschlagnahme (2011) 20: 340-2. doi: 10.1016/j.Beschlagnahme.2010.12.007
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
11. Marson AM, Bucciantini E, Gentile E, Geda C. Neuroakanthozytose: klinische, radiologische und neurophysiologische Befunde in einer italienischen Familie. Neurologie. (2003) 24:188–9. doi: 10.1007/s10072-003-0123-1
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
12. Kurosawa Y., Nakamura M., Ichiba M., Matsuda M., Mizuno E., Kato M., et al. Choreinmangel führt zur Hochregulation von Gephyrin und GABA (A) -Rezeptor. In: Biochem Biophys Res Commun. (2006) 351:438–42. Ursprungsbezeichnung: 10.1016/j.bbrc.2006.10.070
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
13. Ehrlich DJ, Walker RH. Funktionelle Neuroimaging und Chorea: eine systematische Überprüfung. J Clin Mov Disord. (2017) 4:8. ust-IDNR.: 10.1186/s40734-017-0056-0
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
14. Connolly BS, Hazrati L-N, Lang AE. Neuropathologische Befunde bei Chorea-Akanthozytose: Neue Erkenntnisse über Mechanismen, die Parkinsonismus und Krampfanfällen zugrunde liegen. In: Acta Neuropathol. (2014) 127:613–5. ust-idnr.: 10.1007/s00401-013-1241-3
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
15. Sorrentino G, De Renzo A, Miniello S, Nori O, Bonavita V. Spätes Auftreten von Akanthozyten im Verlauf der Chorea-Akanthozytose. In: J Neurol Sci. (1999) 163:175–8.
PubMed Zusammenfassung / Google Scholar
16. Bayreuther C, Borg M, Ferrero-Vacher C, Chaussenot A, Lebrun C. Choréo-acanthocytose sans acanthocytes. Revue Neurol. (2010) 166:100–3. doi: 10.1016/j.neurol.2009.03.005
CrossRef Volltext / Google Scholar
17. Es sind keine frei zugänglichen ergänzenden Materialien verfügbar Zitation Lesage S, Drouet V, Majounie E, Jacoupy M, et al. Der Verlust der VPS13C-Funktion beim autosomal-rezessiven Parkinsonismus verursacht eine mitochondriale Dysfunktion und erhöht die PINK1 / Parkin-abhängige Mitophagie. Bin J Hum Genet. (2016) 98:500–13. Ursprungsbezeichnung: 10.1016/j.ajhg.2016.01.014
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
18. Es sind keine frei zugänglichen ergänzenden Materialien verfügbar Zitation Gauthier J, Hempel M, Mencacci NE, Meijer IA, et al. Rezessive Mutationen in >VPS13D verursachen Bewegungsstörungen im Kindesalter. Ann Neurol. (2018) 83:I6. doi: 10.1002/Jahr.25204
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
19. Kurosawa Y., Nakamura M., Ichiba M., Matsuda M., Mizuno E., Kato M., et al. In vivo Verteilung und Lokalisation von Chorein. In: Biochem Biophys Res Commun. (2007) 353:431–5. Ursprungsbezeichnung: 10.1016/j.bbrc.2006.12.059
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
20. Schöls L, Föller M, Schäffer TE, et al. Neuronen, Erythrozyten und darüber hinaus -die vielfältigen Funktionen von Chorein. Neurosignale (2017) 25:117-26. doi: 10.1159/000485457
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
21. Benninger F, Afawi Z, Korczyn AD, Oliver KL, Pendziwiat M, Nakamura M, et al. Krampfanfälle als präsentierendes und prominentes Symptom bei Chorea-Akanthozytose mit c.2343-VPS13A-Genmutation. Epilepsie (2016) 57: 549-56. doi: 10.1111/epi.13318
PubMed Abstract | CrossRef Full Text | Google Scholar