Grenzen in der Pädiatrie
- Einführung
- Langes QT-Syndrom
- LQTS1
- LQTS2
- LQTS3
- LQTS4
- LQTS5
- LQTS6
- LQTS7
- LQTS8
- LQTS9 und LQTS10
- LQTS11
- LQTS12
- LQTS13
- Erworbenes LQTS
- Elektrokardiographische Merkmale in den drei häufigsten Formen des Long-QT-Syndroms
- Genotyp-Phänotyp
- Klinisches Management von LQTS
- LQTS: Saudi Perspective
- Interessenkonflikterklärung
- Danksagung
Einführung
Familiäre oder erbliche Herzrhythmusstörungen machen einen signifikanten Prozentsatz von Herzrhythmusstörungen aus und sind auch ursächlich für den plötzlichen Herztod (SCD) (1, 2). In den letzten zwei Jahrzehnten haben Wissenschaftler und Kliniker enorme Anstrengungen unternommen, um die komplizierten und komplexen Mechanismen angeborener familiärer Arrhythmien zu entschlüsseln (1-8). Um den Mechanismus der Arrhythmogenese zu verstehen, müssen wir die Grundlagen der Herzzellstruktur und ihre elektrophysiologischen Eigenschaften kennen. Herzmyozyten sind die wichtigsten funktionierenden Zellen im Herzen und sie sind weitgehend gekoppelt, so dass sich Impulse schnell und gleichmäßig ausbreiten. Kardiomyozyten sind durch eine spezielle Grenze, die interkalierte Scheibe, voneinander getrennt; Gap Junction Proteine, Herzdesmosomen und Ionenkanäle befinden sich in der interkalierten Scheibe (9). Gap Junctions bestehen aus dicht gepackten Connexinen, die den interzellulären Austausch kleiner Moleküle ermöglichen und auch erlauben, die exzitatorischen Ströme von einer Zelle zu ihrer benachbarten Zelle zu fließen. Desmosomen sind zusammen mit den Adherens-Übergängen für die mechanischen Anhaftungen einzelner Kardiomyozyten verantwortlich. Alle diese Komponenten in den interkalierten Scheiben sind sequentiell getrennt und jede Komponente übt ihre einzigartige Funktion aus; Die Störung der einen Komponente beeinflusst die Funktion anderer Komponenten, was das Herz für die Entwicklung von Arrhythmien prädisponiert (9-11). Kardiale Ionenkanäle sind porenbildende Proteinkomplexe, die spannungsgesteuerte und komplex koordinierte Bewegungen von Ionenströmen nach innen und außen durch die Zellmembranen liefern, die für die Erzeugung und Ausbreitung des Herzrhythmus unerlässlich sind. Das Long-QT-Syndrom (LQTS), das Short-QT-Syndrom (SQTS), das Sick-Sinus-Syndrom (SSS), der Herzleitungsdefekt (CCD), BrS, die katecholaminerge polymorphe ventrikuläre Tachykardie (CPVT), das frühe Repolarisationssyndrom (ERS) und das familiäre Vorhofflimmern (AF) sind die derzeit bekannten kardialen Kanalopathien, die aufgrund eines einzelnen oder mehrerer Defekte in den Genen auftreten können, die mit der Erzeugung und Ausbreitung des Herzrhythmus verbunden sind.
In dieser Übersicht werden wir ausschließlich über LQTS-verknüpfte Arrhythmien, ihre Pathophysiologie und das derzeit verfügbare klinische Management berichten. Wir haben Pionierarbeit bei der Aufklärung der genetischen Pathologie bei familiären Herzrhythmusstörungen in Saudi-Arabien geleistet (1, 12-14), wir führen immer noch kardiogenetische Untersuchungen in Saudi-Arabien durch. Wir werden auch die kürzlich veröffentlichten Daten zu LQTS von einem Team des King Faisal Specialist Hospital and Research Centre, Riyadh, hinzufügen (15).
Langes QT-Syndrom
Angeborenes LQTS ist eine Erbkrankheit, die durch Verlängerung des QT-Intervalls im Elektrokardiogramm (EKG) definiert wird. Patienten mit allen Formen von LQTS sind prädisponiert für ventrikuläre Tachyarrhythmie, Torsades de pointes (TdP), die zu rezidivierenden Synkopen oder SCD führen. In vielen Fällen könnte Synkope oder plötzlicher Tod die erste und einzige Manifestation sein. LQTS betrifft schätzungsweise 1 von 2.000 Menschen weltweit (16). Das Markenzeichen des LQTS ist die Verlängerung des QT-Intervalls im EKG (korrigiert um die Herzfrequenz, d. H. QTc). Normalwerte von QTc sind 440 ms bei Männern und 450 ms bei Frauen. Bei Kindern sind alters- und geschlechtsabhängige Werte relevant. Die jüngsten Konsensempfehlungen für die LQTS-Diagnose lauten wie folgt (17, 18):
1. LQTS wird diagnostiziert:
a. Bei Vorliegen eines LQTS-Risikoscores ≥3,5 in Abwesenheit einer sekundären Ursache für eine QT-Verlängerung und/oder
b. Bei Vorliegen einer eindeutig pathogenen Mutation in einem der LQTS-Gene oder
c. In Gegenwart eines QT-Intervalls, korrigiert für die Herzfrequenz unter Verwendung der Bazett-Formel (QTc) ≥500 ms im wiederholten 12-Kanal-Elektrokardiogramm und in Abwesenheit einer sekundären Ursache für eine QT-Verlängerung.
2. LQTS kann in Gegenwart eines QTc zwischen 480 und 499 ms in wiederholten 12-Kanal-EKGs bei einem Patienten mit ungeklärter Synkope in Abwesenheit einer sekundären Ursache für QT-Verlängerung und in Abwesenheit einer pathogenen Mutation diagnostiziert werden.
Patienten mit einer QTc-Dauer ≥500 ms hatten eine signifikant höhere kumulative Wahrscheinlichkeit, ihre erste Synkope zu erleiden, als Patienten mit einer QTc-Dauer < 500 ms von der Geburt bis zum Alter von 20 Jahren (19). Bei Patienten mit 2, 3 und 4 Synkopen-Episoden war das Risiko einer nachfolgenden Synkopen-Episode bei Patienten mit enger oder verlängerter QTc-Dauer praktisch identisch (19). Die molekulare Basis von LQTS ist heterogen und bisher wurden Mutationen in 13 verschiedenen Genen im Zusammenhang mit LQTS beschrieben (1-8, 19, 20). In einem dieser 13 Gene (1-8, 19, 20) findet sich in der Regel bei 70% der LQTS-Patienten ein genetischer Defekt. Unter den derzeit bekannten 13 verschiedenen Arten von LQTS sind die häufigsten LQTS1, LQTS2 und LQTS3 aufgrund von Defekten in kardialen Ionenkanalgenen, KCNQ1, KCNH2 und SCN5A. Neunzig Prozent aller LQTS-Kausalmutationen finden sich in diesen drei Genen (1-8, 19, 20). Mutationen in den verbleibenden 10 Genen sind selten und machen nur ∼10% aller derzeit bekannten LQTS-Mutationen aus.
Das Long-QT-Syndrom ist normalerweise eine autosomal dominante Erkrankung, aber gelegentlich konnten bei 5-10% der Patienten mit LQTS mehrere Mutationen in einem einzigen Gen oder in verschiedenen Genen gefunden werden (21, 22). Patienten mit mehreren Mutationen könnten im Vergleich zu Patienten mit einer einzigen Mutation eine längere QTc aufweisen, und solche Patienten haben auch ein ∼ 3,5-fach erhöhtes Risiko für lebensbedrohliche kardiale Ereignisse (21, 22)
LQTS1
Mutationen im KCNQ1-Gen sind die häufigste Form aller LQTS und werden als LQTS Typ 1 (LQTS1) bezeichnet. Fünfzig Prozent aller LQTS-Kausalmutationen finden sich im KCNQ1-Gen (20). KvLQT1 (auch Kv7.1 genannt) ist ein Protein, das durch das KCNQ1-Gen hergestellt wird, und das Protein bildet Tetramere im endoplasmatischen Retikulum in den Zellen, die sich mutmaßlich mit dem Protein minK (kodiert durch KCNE1) zusammensetzen und dann zur Plasmamembran der Herzmyozyten transportiert werden, wo sie einen langsam aktivierenden Strom vermitteln, der die Repolarisation des Aktionspotentials in Herzgeweben beschleunigt. Dieser Strom ist als IKs bekannt (Abbildung 1). LQTS1- und KCNQ1-Mutationen sind meist Missense-Mutationen und können in seltenen Fällen Frameshift-Mutationen in der C-terminalen Region sein (23). Herzrhythmusstörungen bei KCNQ1-Mutationsträgern werden durch adrenerge Antriebe ausgelöst, z. B. emotionaler Stress, körperliche Anstrengung, Tauchen, Schwimmen (24-26). Patienten mit Mutationen an den Transmembrandomänen von KvLQT1 haben ein höheres Risiko für LQTS-bedingte kardiale Ereignisse und eine höhere Empfindlichkeit gegenüber sympathischer Stimulation (26, 27). Patienten mit Missense-Mutationen haben ein erhöhtes Risiko im Vergleich zu Patienten mit Non-Sense- oder Truncing-Mutationen (27, 28). Die Variation der 3′-UTR im KCNQ1-Gen beeinflusst auch die Arrhythmieanfälligkeit signifikant, vermutlich durch Beeinflussung der Expression des Gens (29). Patienten mit Arrhythmien aufgrund von KCNQ1-Mutationen sprechen recht gut auf β-Blocker an, aber einige Patienten könnten immer noch weniger auf dieses Medikament ansprechen oder sogar resistent sein. In einem kürzlich erschienenen Artikel, Barsheshet et al. (30) behaupteten, dass die Patienten mit Mutationen außerhalb der zytoplasmatischen Schleife (c-Loop) im KvLQT1 weniger auf β-Blocker ansprechen.
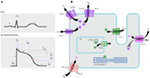
Abbildung 1. Ionenströme, die zum ventrikulären Aktionspotential beitragen (A) und schematische Darstellung eines Kardiomyozyten, der (nur) diejenigen Proteine zeigt, die an der Pathogenese vererbter Arrhythmiesyndrome beteiligt sind (B). In (A) wird das Aktionspotential mit seinem ungefähren Zeitpunkt der Aktion während des EKG ausgerichtet. In (B) ist Ankyrin-B, ein Adapterprotein, das am Long-QT-Syndrom Typ 4 beteiligt ist, nicht dargestellt.
Homozygote oder zusammengesetzte heterozygote Mutationen im KCNQ1-Gen, die die rezessive Form der Krankheit, das Jervell- und Lange-Nielsen-Syndrom (JLNS), Typ 1 (31), verursachen könnten, sind ziemlich selten. JLNS-Patienten leiden ebenfalls an schweren Herzrhythmusstörungen und Taubheit (31, 32). Patienten mit KCNQ1-Mutationen, die zu JLNS führen, haben normalerweise keine funktionellen IKs (28, 31-33). Es ist möglich, dass einige Patienten trotz homozygoter oder zusammengesetzter heterozygoter Mutationen in KCNQ1 keine Taubheit aufweisen, solche Fälle werden als autosomal rezessiv bezeichnet LQTS1 (13, 32). Bei diesen Patienten könnte noch eine geringe Menge an funktionellem IKs-Strom (< 10% des gesamten IKs) vorhanden sein, wodurch die Hörfunktion erhalten bleibt, aber ihre Herzrhythmusstörungen sind ebenso schwerwiegend wie bei JLNS-Patienten (13, 32).
LQTS2
Diese Art von LQTS ist ebenso häufig wie LQTS1 und macht 35-40% der LQTS-Patienten mit einer nachweisbaren Mutation aus (1, 20, 24, 25). KCNH2 kodiert für das HERG-Protein (Kv11.1), das die α-Untereinheit des schnell aktivierenden verzögerten Gleichrichters K + current (IKr) ist. Pathogene Mutationen in diesem Gen, die die Kv11.1-Kanalfunktion reduzieren, verlängern die Dauer des QT-Intervalls (Abbildung 1) und sind ursächlich für LQTS2. Neunundzwanzig Prozent der Synkopenattacken in LQTS2 treten während Ruhe / Schlaf auf und nur 13% der Synkopenattacken treten während des Trainings auf (25, 34). Plötzliche überraschende Geräusche, z. B. Wecker klingelt, Türklingeln, Telefon klingelt, in der Regel auslösen synkopalen Attacken bei diesen Patienten (24, 34). Patienten mit Mutationen in der porenbildenden Region des Kv11.1 (kodiert durch das KCNH2-Gen) sind im Vergleich zu Patienten mit Mutationen außerhalb der Porenregion anfällig für ein hohes Risiko für Arrhythmie-bedingte kardiale Ereignisse (35). Bei Neugeborenen ist der 2: 1 atrioventrikuläre (AV) Block bevorzugt mit KCNH2-Mutationen assoziiert (36). Ein vollständiger AV-Block, der durch LQTS kompliziert wurde, wurde auch bei 17% der erwachsenen Patienten mit einer Mutation im KCNH2-Gen gefunden (37). Homozygote Mutationen in KCNH2 sind selten und wenn vorhanden, Patienten leiden an einer schweren Form von LQTS, mit 2:1 AV-Block und schwere ventrikuläre Arrhythmien, sowohl im intrauterinen Stadium als auch nach der Geburt (11, 38-40). Zusätzlich zu den zahlreichen Mutationen, die in der LQTS2-Pathologie beschrieben werden, könnten auch häufige polymorphe Varianten im KCNH2-Gen die Schwere der Erkrankung modulieren. Ein interessantes Beispiel ist der K897T-Polymorphismus (SNP) in KCNH2, der in 33% der Allgemeinbevölkerung vorhanden ist (41). Es wurde berichtet, dass K897T die Pathogenität der KCNH2-Mutationen verschlimmert (42, 43).
LQTS3
Nav1.5 ist die porenbildende α-Untereinheit des spannungsabhängigen kardialen Na + -Kanals, es ist ein integrales Membranprotein, das vom SCN5A-Gen codiert wird und an der Initiierung und Leitung von kardialen Aktionspotentialen beteiligt ist. Kardiale Na + -Kanäle bestehen aus einer porenbildenden α-Untereinheit (codiert durch SCN5A) und einer oder mehreren β-Hilfsuntereinheiten.
LQTS3 wird durch Funktionsgewinn verursacht Mutationen, die die schnelle Inaktivierung der α-Untereinheit stören (Abbildung 1), und eine Mutation im SCN5A-Gen wurden bei < 10% aller LQTS-Patienten mit einer Mutation beschrieben (1, 20, 24). LQTS3-Patienten erleben die Mehrheit (39%) ihrer kardialen Ereignisse während des Schlafes / der Ruhe (25), und über ∼13% der Ereignisse wurde berichtet, dass sie während des Trainings auftreten (25). In mehreren Fällen zeigte eine einzelne SCN5A-Mutation zwei oder sogar drei verschiedene Phänotypen von Arrhythmien in derselben Familie, z. B. LQTS, BrS oder CCD (44-47). Männliche Patienten mit einer LQTS3-Mutation könnten Symptome viel früher entwickeln als weibliche Patienten (48). Sowohl heterozygote als auch homozygote SCN5A-Mutationen wurden in LQTS3 mit funktionellem 2:1 AV-Block beschrieben (49). Wir haben kürzlich eine iranische Familie mit der 1507_1509delQKP-Mutation in mehreren Familienmitgliedern gefunden, in denen Patienten LQTS und CCD kombiniert hatten (Daten nicht gezeigt). Diese Mutation wurde auch bei Patienten aus anderen Ländern berichtet, was darauf hindeutet, dass es sich um eine wiederkehrende Hot-Spot-Mutation handelt . Mutationen mit solchen Verlust- und Funktionsgewinn-Eigenschaften während verschiedener Phasen des Aktionspotentials wurden auch in 1493delK- und 1795insD-Mutationen berichtet (44, 51).
Gelegentlich könnte ein SNP auch eine pathogene Wirkung auf seine Träger ausüben. S1103Y ist eine häufige Variante des SCN5A-Gens, die bei 13% der Afroamerikaner vorkommt (52). Träger mit dieser Variante haben ein erhöhtes Risiko für Arrhythmien und plötzlichen Kindstod (SIDS) (52).
LQTS4
LQTS4 stellt die erste Nicht-Kanal-Form von LQTS dar. Eine Mutation im ANK2, einem Adapterprotein, führt zu einer intrazellulären Calciumüberladung, die zum LQTS4 beiträgt (53, 54). Zusätzlich zur QT-Verlängerung könnten Patienten mit diesem Syndrom eine Sinusbradykardie, paroxysmales Vorhofflimmern und CPVT aufweisen (54). Die Wirkung der ANK2-Mutationen kann moderat bis schwerwiegend sein und die klinischen Ausprägungen hängen vom Schweregrad der Mutation ab.
LQTS5
Mutationen in KCNE1 sind mit dem LQTS5 assoziiert (Abbildung 1) (55, 56). Heterozygote KCNE1-Mutationen reduzieren IKs, indem sie einen dominanten negativen Effekt auf das begleitende normale Allel ausüben, und führen zu einer verzögerten kardialen Repolarisation (Abbildung 1), die für ein erhöhtes Risiko für Arrhythmien verantwortlich ist (6). Patienten mit homozygoten KCNE1-Mutationen leiden an JLNS (Typ 2) (57, 58).
D85N ist ein Polymorphismus im KCNE1-Gen, der in 0 vorhanden ist.7-1% der Gesamtbevölkerung (41). In einer Studie von Nishio et al. (59) wurde der D85N-Polymorphismus häufiger bei den LQTS-Patienten gefunden, was ihn zu einem Risikogenotyp in der LQTS-Pathologie macht (möglicherweise nur in der asiatischen Bevölkerung). In Europa wurde D85N bei 5% der erworbenen LQTS (aLQTS) -Patienten (in einer Kohorte von 32 Patienten) berichtet, bei denen TdP aufgetreten war (60).
LQTS6
Das KCNE2-Gen kodiert für MinK-related Peptide 1 (MiRP1), eine mutmaßliche β-Untereinheit des kardialen Kaliumkanals IKr (Abbildung 1). Mutationen im KCNE2-Gen könnten auch zu Defekten in der schnell aktivierenden Komponente des verzögerten Gleichrichter-Kaliumstroms (IKr) führen, pathologische Grundlage von LQTS6 (61). Auditive/akustische reiz wie wecker lärm, türklingel etc. könnte synkopale Angriffe in KCNE2-Mutationsträgern hervorrufen, ähnlich der KCNH2-Mutation (62).
LQTS7
Dieses Syndrom ist auch als Andersen–Tawil-Syndrom (ATS) bekannt. ATS ist eine seltene Erkrankung, die sich durch gelegentliche Synkope und Herzstillstand manifestiert. Zu den EKG-Merkmalen gehören eine leichte Verlängerung des QT-Intervalls, abnormale U-Wellen, häufige ventrikuläre Ektopie, bidirektionale ventrikuläre Tachykardie (VT) und polymorphe VT. Dieses Syndrom weist auch extrakardiale Merkmale auf, z. B. periodische Skelettmuskellähmungen und Entwicklungsprobleme wie Gaumenspalte, niedrig angesetzte Ohren, Kleinwüchsigkeit und Entwicklungsdefekte in den Gliedmaßen (63). Es wurde berichtet, dass die Mehrheit der klinisch diagnostizierten ATS-Patienten eine Mutation in KCNJ2 aufwies (63). KCNJ2 kodiert für eine porenbildende Untereinheit von nach innen gleichrichtenden Kaliumkanälen (IK1) (Abbildung 1) (64, 65).
LQTS8
Patienten, die auch als Timothy-Syndrom (TS) bekannt sind, zeigen bei ihren EKGs eine schwere QT-Verlängerung, die in 100% der Fälle mit Syndaktylie, Kahlheit bei der Geburt und kleinen Zähnen und weniger penetrant einhergeht Herzstrukturfehlbildungen, geistige Behinderung, Autismus und gesichtsdysmorphe Merkmale (66). Es gibt zwei Subtypen: TS1 (klassisch) und TS2 (seltene Form).
TS2 ist kardiologisch schwerer als TS1 (66, 67). TS2-Patienten fehlt auch Syndaktylie (67). Mutationen in der α-1-Untereinheit des L-Typ-Calcium-Stroms (ICa-L), das für das Gen CACNA1C kodiert, führen zu beiden Formen von TS (LQTS8). Es gibt alternativ gespleißte, sich gegenseitig ausschließende Exon-8 im CACNA1C-Gen, zur Verdeutlichung werden sie als Exon-8 und Exon-8A bezeichnet. In Herz und Gehirn, wo das CACNA1C überwiegend exprimiert wird, findet sich Exon-8 in ∼80% der mRNA-Transkripte und Exon-8A ist vorhanden ∼20% Transkripte (66). Die G406R-Mutation in Exon-8A ist ursächlich für die klassische Form von TS (TS1) und G402S in Exon-8 wurde bei Severer in TS2 berichtet. Neu in dieser Liste ist die Ala1473Gly-Mutation, die in einem TS-Säugling mit weiter erweitertem Phänotyp beschrieben wurde (68). Alle Mutationen waren entweder de novo oder Mosaik im Elternteil, und alle resultierten aus der Funktion des ICa-L-Kanals (66-69).
LQTS9 und LQTS10
Mutationen in CAV3 oder SCN4B führen zu einem Funktionsgewinn in der späten INa und verursachen einen LQTS3-ähnlichen Phänotyp (70-74). Sie sind als LQTS9 (assoziiert mit CAV3-Mutation) und LQTS10 (assoziiert mit SCN4B-Mutation) bekannt.
Caveolae werden von Engelman et al. (72) als “kleine Höhlen” in der Plasmamembran. Sie sind kleine unbeschichtete Gruben und gelten als Ort wichtiger dynamischer und regulatorischer Ereignisse an der Plasmamembran (72, 73). Caveoline sind die Hauptproteine in den Caveolen, und Caveolin-3 (kodiert durch das Gen CAV3) kommt spezifisch in Kardiomyozyten und Skelettmuskelzellen vor. Es wurde speziell berichtet, dass mehrere kardiale Ionenkanäle in den Kaveolen lokalisiert sind, die aus Herzmyozyten extrahiert wurden, die mit Caveolin-3 angereichert sind (72, 73). Zusätzlich sind Komponenten der β-adrenergen Rezeptor-Signalkaskade auch in caveolae-angereicherten Membranen vorhanden (72, 73).
SCN4B kodiert für NaVß4, das eine zusätzliche β-Untereinheit des kardialen Natriumkanals ist. Bisher wurde nur eine Mutation (L179F) in diesem Gen in einer mexikanischen Familie mit mehreren betroffenen Familienmitgliedern berichtet (74). Es wurde festgestellt, dass eine Mutation in diesem Gen zu einem Funktionsgewinn des Nav1.5-Stroms führt (74).
LQTS11
Im Herzen wird die sympathische Regulation der kardialen Aktionspotentialdauer (APD) durch die Aktivierung des β-adrenergen Rezeptors (β-AR) vermittelt, was die Assemblierung von AKAP9 (Yotiao) mit der α-Untereinheit (KvLQT1) des IKs-Kanals erfordert. Mutation in AKAP9 verursacht LQTS11 (75). Bisher wurde nur über eine Mutation, S1570L, in AKAP9 berichtet (75).
LQTS12
Mutationen im α-1-Syntrophin (SNTN1)-Gen sind ursächlich für LQTS12. Die Mutation in diesem Gen führt zu einem Funktionsgewinn des kardialen Natriumkanals (Nav1.5), der die pathologische Grundlage von LQTS12 darstellt (76).
LQTS13
Eine proteingekoppelte, nach innen korrigierende Kaliumkanal-Untereinheit (Kir3.4) wird durch das KCNJ5-Gen kodiert. Eine Funktionsverlustmutation in diesem Gen könnte LQTS13 verursachen (77). Bisher wurde nur eine Mutation, G387R, in einer chinesischen Familie mit neun Patienten mit dieser Mutation beschrieben. Eine reduzierte Plasmamembranexpression von Kir3.4 wurde als Pathologie von LQTS bei den Patienten vorgeschlagen.
Erworbenes LQTS
Neben dem angeborenen LQTS gibt es auch eine andere Variante des LQTS, die als aLQTS bekannt ist und durch Faktoren und Substanzen verursacht wird, die den Kaliumfluss verringern und die Repolarisierungsfähigkeit des Myokards beeinträchtigen. Bekannte Erkrankungen sind weibliches Geschlecht, Hypokaliämie und Medikamente, die kardiale Kaliumkanäle hemmen (78-80). Eine Reihe von häufig verschriebenen Medikamenten könnte auch den HERG-Kanal (Kv11.1, ein Protein, das durch das KCNH2-Gen kodiert wird) bevorzugt binden und blockieren und für aLQTS prädisponieren (79, 80). Kürzlich wurde gezeigt, dass die Blockade von IKs auch zu arzneimittelinduzierten aLQTS beitragen kann, insbesondere wenn die Repolarisationsreserve beeinträchtigt ist (81). Es wurde festgestellt, dass Fluoxetin und Norfluoxetin sowohl in vivo als auch in vitro die IKs-Eigenschaften unterdrücken und zu ausgeprägten LQTS führen (81). Es wurde berichtet, dass Polymorphismen, D85N in minK (Gen KCNE1), T8A, Q9E in MiRP1 (Gen KCNE2), die mutmaßliche β-Untereinheiten der IKs- und IKr-Kanäle sind, aLQTS verursachen (82, 83). Autoimmun-LQTS wurde auch bei einem Patienten mit IgG-haltigen Anti-HERG-Antikörpern berichtet (84).
Elektrokardiographische Merkmale in den drei häufigsten Formen des Long-QT-Syndroms
Typische ST-T-Wellenmuster sind bei der Mehrzahl der genotypisierten LQTS-Patienten vorhanden und können zur Identifizierung von LQTS1-, LQTS2- und möglicherweise LQTS3-Genotypen verwendet werden (85, 86).
Die LQTS1-Form des LQTS ist mit einer breiten T-Welle ohne Verkürzung des QT-Intervalls bei Belastung assoziiert (Abbildung 2A). LQTS2 ist mit T-Wellen niedriger Amplitude, oft bifid, assoziiert (Abbildung 2B). LQTS3 ist mit einem langen iso-elektrischen Segment und einer schmalen, hohen T-Welle assoziiert (Abbildung 2C). Die Abhängigkeit des TdP-Beginns bei angeborenen LQTS ist genotypspezifisch und überwiegt bei LQTS2, fehlt jedoch bei LQTS1 fast (87).

Abbildung 2. Elektrokardiogramm-Aufnahmen von Patienten mit LQTS1, LQTS2 und LQTS3. (A) 12-Kanal-EKG eines 18-jährigen Mannes mit einer KCNQ1-Mutation. Das QT-Intervall ist verlängert (QTc = ± 500 ms). Das ST-Segment hat eine breite Basis und eine relativ große Amplitude. Leitungsintervall ist normal (Standardkalibrierung). (B) 12-Kanal-EKG eines 14-jährigen Mädchens mit einer KCNH2-Mutation. Das QT-Intervall ist verlängert (QTc ± 520 ms). Das ST-Segment ist in der Leitung V3 eingekerbt und hat eine relativ geringe Amplitude in den Extremitätenleitungen. Leitungsintervall ist normal (Standardkalibrierung). (C) 12-Kanal-EKG eines 12-jährigen Jungen mit einer SCN5A-Mutation. Das QT-Intervall ist verlängert (QTc ± 600 ms). Das ST-Segment hat ein langes (fast) iso-elektrisches Segment mit einer großen, scharfen und schmalen T-Welle. Leitungsintervall ist normal (Standardkalibrierung).
Obwohl Muster auf einen spezifischen Genotyp des LQTS hindeuten können, wurden häufig Ausnahmen gefunden.
Genotyp-Phänotyp
Das Long-QT-Syndrom ist eine autosomal dominante Erkrankung. Genotyp- und Phänotypanalysen unter den heterozygoten Mutationsträgern wurden bei den LQTS1-, LQTS2- und LQTS3-Patienten recht umfangreich durchgeführt. Im Kindesalter ist das Risiko für kardiale Ereignisse bei LQTS1-Männern signifikant höher als bei LQTS1-Frauen, während bei LQTS2- und LQTS3-Patienten keine signifikanten geschlechtsspezifischen Unterschiede im Risiko für kardiale Ereignisse beobachtet wurden (88-91). Im Erwachsenenalter (auch nach dem 40. Lebensjahr) könnten LQTS1- und LQTS2-Frauen ein signifikant höheres Risiko für kardiale Ereignisse haben als die jeweiligen Männer (88-91). Im Allgemeinen scheint die Letalität kardialer Ereignisse bei LQTS3-Patienten vorherrschend zu sein als bei LQTS1- und LQTS2-Patienten (91). Frauen mit LQTS haben ein reduziertes Risiko für kardiale Ereignisse während der Schwangerschaft, aber das Risiko steigt während der 9-monatigen postpartalen Phase, insbesondere bei Frauen mit Mutation im KCNH2-Gen (92).
Der plötzliche Herztod bei Kindern könnte auch durch Mutationen in den kardialen Ionenkanalgenen verursacht werden (93-96). Etwa 28% der Kinder mit einer ungeklärten SCD (zwischen 1 und 18 Jahren, Durchschnittsalter: 12,3 ± 3,8 Jahre) waren Träger von Mutationen in LQTS-Kausalgenen (97). Bei SIDS schienen Mutationen in SCN5A vorherrschend zu sein (98, 99), aber auch Mutationen in KCNQ1, KCNH2, KCNE2 und CAV3, SCN4B und SCN3B wurden gefunden (100, 101). Intrauterine fetale Todesfälle wurden auch aufgrund von Defekten in kardialen Ionenkanalgenen berichtet (12, 102).
Klinisches Management von LQTS
Das Absetzen aller Medikamente, von denen bekannt ist, dass sie das QT-Intervall verlängern, sowie die Korrektur von Elektrolytstörungen und / oder auslösenden Stoffwechselzuständen sollten im Mittelpunkt der Behandlung von (Neu-) LQTS-Patienten stehen. Die Symptome bei LQTS sind häufig adrenerg vermittelt, daher wird im Allgemeinen empfohlen, die Teilnahme der Patienten an sportlichen Aktivitäten einzuschränken (24, 25). Die Hauptstütze der klinischen Therapie für das LQTS ist die β-Blockade. Üblicherweise werden langwirksame Präparate von Propranolol, Nadolol und Metoprolol verwendet, und ihre Wirksamkeit bei der β-Blockade wird durch Abstumpfung der Belastungsherzfrequenz (z. B. durch >20%) (24, 25). Unter allen β-Blockern werden Propranolol und Nadolol bei symptomatischen Patienten als überlegen gegenüber Metoprolol angesehen (103). Darüber hinaus kann β-Blockade auch als prophylaktische Behandlung in stillen Mutationsträgern verwendet werden, um SCD zu reduzieren (24). In einer Studie von Barsheshet et al. (30) zeigten Patienten mit c-Loop-Missense-Mutationen im KCNQ1-Gen ein hohes Risiko für lebensbedrohliche kardiale Ereignisse und sie hatten signifikante Vorteile von der Behandlung mit β-Blockern. Da Frauen mit LQTS2 während der 9-monatigen postpartalen Phase ein erhöhtes Risiko haben, sollten β-Blocker verschrieben werden, um kardiale Ereignisse während dieser Hochrisikoperiode zu reduzieren (92). Ein implantierbarer Kardioverter-Defibrillator (ICDs) kann bei Patienten mit rezidivierender Synkope trotz β-Blocker-Therapie oder bei Patienten mit hohem Risiko für einen Herzstillstand (z., symptomatische LQTS2 und LQTS3 mit dokumentierter QTc-Verlängerung). Die sympathische Denervation des linken Herzens (LCSD) wird für LQTS-Patienten mit hohem Risiko empfohlen, bei denen ein ICD kontraindiziert oder abgelehnt ist und β-Blocker entweder nicht wirksam, nicht toleriert, nicht akzeptiert oder kontraindiziert sind (18, 104). JLNS-Patienten haben in der Regel ein QTc > 500 ms und sind ebenfalls einem hohen Risiko ausgesetzt, β-Blocker haben eine unzureichende Wirksamkeit bei solchen Patienten, bei denen eine frühzeitige Therapie mit ICD empfohlen wurde (18, 31).
LQTS: Saudi Perspective
Der erste Bericht über LQTS aus Saudi-Arabien wurde 1993 vom Riyadh Armed Forces Hospital veröffentlicht (105). Vier Säuglinge und Kleinkinder zwischen 6 und 48 Monaten mit wiederkehrenden Anfällen in der Vorgeschichte aus einer einzigen Familie wurden als LQTS diagnostiziert (105). Die Familiengeschichte ergab, dass zwei weitere Familienmitglieder ähnliche Episoden plötzlichen Bewusstseinsverlusts hatten und drei Familienmitglieder plötzlich starben (105). In allen Fällen war die Erstdiagnose Epilepsie (105). Einige Jahre später wurden zwei sporadische Fallberichte mit einer vergleichsweise schwereren Variante des Neugeborenen-LQTS in Kombination mit einem 2: 1-AV-Block berichtet (106, 107). Alle diese veröffentlichten klinischen Berichte enthielten keine genetischen Befunde, die die Pathophysiologie von LQTS in ihnen erklären könnten (105-107).
Wir haben zum ersten Mal genetische Defekte als pathologische Grundlage von LQTS bei ähnlichen Patienten aus Saudi-Arabien gemeldet. Wir haben sechsköpfige Familien mit Synkopen in der Vorgeschichte und plötzlichen unerklärlichen Todesfällen von Fötus, Neugeborenen und Kindern untersucht (12-14). Autosomal-rezessives LQTS1 wurde bei Kindern aus zwei Familien diagnostiziert (Abbildung 3). Autosomal-rezessives LQTS2 wurde in zwei Familien diagnostiziert (Abbildung 4). In einer Familie wurde bei einer Patientin autosomal dominantes LQTS2 diagnostiziert (Abbildung 5), die Patientin hatte synkopale Anfälle während der postpartalen Erholungsphase im Krankenhaus, was bei Frauen mit KCNH2-Mutationsträgern sehr häufig vorkommt. Bei allen unseren Patienten führte die Identifizierung pathogener Mutationen in den LQTS-kausalen kardialen Ionenkanalgenen zu einer bestätigten klinischen Diagnose, die vor der Überweisung an uns fälschlicherweise als epileptische Krämpfe diagnostiziert wurden (13, 14). (15) vom King Faisal Specialist Hospital und Forschungszentrum berichteten über eine LQTS1-kausale KCNQ1-Mutation, H258P, in einer großen Familie mit 12 betroffenen Personen. Nur zwei Träger waren symptomatisch, ihre QTc waren > 500 ms, und β-Blocker unterdrückten die klinischen Symptome bei einem Patienten und der zweite symptomatische Patient benötigte einen ICD.
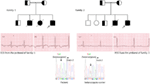
Abbildung 3. Stammbaumzeichnung der Familie-1 und 2 mit autosomal rezessivem LQTS1, Probanden sind mit einem Pfeil dargestellt. EKGs von den Probanden in zwei Familien sind in der Mitte gezeigt, mit einem QTc von 557 bzw. 529 ms. Intronische Mutation, c.387 -5 T > A im KCNQ1-Gen wurde bei den Patienten gefunden (unten gezeigt). Die Mutation wurde mit einem Pfeil angezeigt. Nicht gefüllte Kreise und Quadrate sind keine Träger für die Mutation. Betroffene Personen werden als gefüllte Kreise (weiblich) und Quadrate (männlich) dargestellt. Halbgefüllte Quadrate und Kreise sind Individuen mit heterozygoter Mutation. Verstorbene Personen werden durch Schrägstriche angezeigt, Probanden werden durch einen Pfeil angezeigt und die blutsverwandte Ehe wird durch angezeigt = Die Exon – Intron-Grenze wird durch eine gepunktete Linie mit Pfeil in Richtung Exon angezeigt.
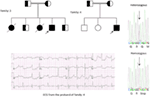
Abbildung 4. Stammbaumzeichnung der Familie-3 und 4 mit autosomal rezessivem LQTS2. Das EKG des Probanden aus Familie-4 ist unterhalb der Stammbaumzeichnung dargestellt, die Sinustachykardie, fast vollständigen AV-Block, breiten komplexen Fluchtrhythmus mit sehr langen QTc-Intervallen (QT > 600 ms) zeigt. Mutation, c.3208 C > T (p.Q1070X) im KCNH2-Gen wurde bei den Patienten gefunden (rechts dargestellt), markiert mit einem Pfeil.
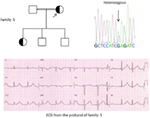
Abbildung 5. Oben links: Stammbaum der Familie-5. Betroffene Person wird durch einen ausgefüllten Kreis angezeigt (weiblich) Proband wird durch einen Pfeil angezeigt. 12-Kanal-EKG des Probanden (I:2). EKG zeigt eine hohe Spitze, breit angelegte, große Amplitude T-Welle, QTc-Intervall beträgt 580 ms. Screening des KCNH2-Gens zeigt Substitution von Nukleotid “G” für ein “A” (c.2362G > A, Pfeil markiert), was zu Aminosäuresubstitution führt, p.E788K.
Genetische und klinische Befunde in unserer Studie (12-14) aus Saudi-Arabien sind aus mehreren Gründen sehr faszinierend: (1) Insgesamt haben wir sechs Familien untersucht, darunter vier waren homozygot / Verbindung heterozygot für die Mutationen und die Mutationen stammten aus einer Ahnenquelle; (2) Alle Mutationen in LQTS waren neuartig und wurden nur in diesen arabischen Familien berichtet; (3) Aufgrund der Homozygotie oder zusammengesetzten Heterozygotie für die Mutationen waren die klinischen Phänotypen auch in unseren untersuchten Familien schwerwiegend (12-14). Wir schlugen vor, dass die genetischen und phänotypischen Beobachtungen auf die extrem hohe Rate blutsverwandter Ehen in Saudi-Arabien zurückzuführen sind (108, 109). Unsere Studie lieferte den ersten wissenschaftlichen Beweis für die Rolle der Blutsverwandtschaft bei der Ausübung einer Schlüsselrolle bei ungeklärten Herzrhythmusstörungen und SCDs bei Kindern und Jugendlichen in Saudi-Arabien (12-14). Wir haben auch gezeigt, dass sich die KCNQ1-Mutation c.387 -5 T > A (NM_000218) (Abbildung 3) in der Provinz Assir in Saudi-Arabien von einem gemeinsamen Vorfahren während mehrerer Generationen aufgrund der hohen Inzidenz von blutsverwandten Ehen verbreitet hatte . Bisher ist dies die häufigste LQTS1-Kausalmutation in der saudi-arabischen Bevölkerung, die auch im King Faisal Specialist Hospital und im Khamis Mashayt Military Hospital beobachtet wurde (unveröffentlicht). Ein ähnliches Ergebnis wurde auch für die LQTS2-Kausalmutation erhalten, c.3208 C > T (p.Q1070X) (Abbildung 4) im KCNH2-Gen (12, 14), das ebenfalls eine Gründermutation in Saudi-Arabien ist und über viele Generationen in der Region Assir segregiert wurde (siehe Karte, Abbildung 6). Wir würden vorhersagen, dass es eine beträchtliche Anzahl von Individuen mit den genannten Vorfahren / Gründer-Mutationen (sowohl in KCNQ1 und KCNH2 und anderen Genen) in dieser Region und auch in den großen Städten wie Riad, Jeddah und Dammam aufgrund der städtischen Migration gibt. Mehr Gründer- oder Ahnenmutationen, die für LQTS pathogen sind, sind in anderen Provinzen Saudi-Arabiens aufgrund der hohen Rate an blutsverwandten Ehen sehr wahrscheinlich.
Abbildung 6. Karte von Saudi-Arabien (mit freundlicher Genehmigung: Wikipedia). Die Assir-Region ist mit einer dicken Linie markiert, in der wir die Gründermutationen in den Genen KCNQ1 und KCNH2 gefunden haben, die in Familie 1-4 beschrieben sind.
Da LQTS eine autosomal dominante Erkrankung ist, erwarteten wir, überwiegend heterozygote Mutationsträgerpatienten zu beobachten. In unserer Studie identifizierten wir jedoch hauptsächlich Patienten mit rezessiven Mutationen (12-14). Offensichtlich werden diese Patienten zuerst symptomatisch und wurden überwiegend an das Prince Sultan Cardiac Centre überwiesen, um sie unter unsere Aufmerksamkeit zu bringen. Nichtsdestotrotz deuten die Ergebnisse unserer Untersuchungen darauf hin, dass rezessive LQTS bei Kindern tödliche klinische Phänotypen haben können und in Saudi-Arabien nicht ungewöhnlich sind (12-14). Da die heterozygoten Mutationsträger auch anfällig für Arrhythmien und deren Komplikationen sind, sollte eine konzertierte Initiative ergriffen werden, um die örtlichen Allgemeinärzte, Kardiologen und klinischen Genetiker auf eine gemeinsame Plattform zu bringen, um die gefährdeten Personen zu identifizieren. Darüber hinaus haben wir derzeit auch keine genetischen Informationen für andere familiäre Arrhythmiestörungen, z. B. CPVT, SQTS, BrS, AF usw. Spezialisierte kardiogenetische Zentren sollten die Initiative ergreifen, um nach genetischen Defekten und Mutationen zu suchen und Genotyp-Phänotyp-Studien bei allen Formen erblicher Arrhythmien durchzuführen. Es sollte auch berücksichtigt werden, dass nicht alle genetischen Arrhythmien eine Familienanamnese haben, da in vielen Fällen Arrhythmie-ursächliche Mutationen de novo-Ursprungs sind, dh der Proband ist der erste Patient in dieser Familie mit der Mutation und er / sie ist die Quelle, um die Mutation in nachfolgenden Generationen zu übertragen (110, 111). Aufgrund der sehr hohen Rate an blutsverwandten Ehen in Saudi-Arabien, Wir erwarten viel mehr Gründermutationen, die hierzulande eine entscheidende Rolle bei angeborenen Herzrhythmusstörungen spielen. Die Identifizierung dieser Gründermutationen sollte unsere erste und wichtigste Aufgabe sein, die es uns erleichtern würde, eine wirksame voreheliche und auch präsymptomatische genetische Beratung in diesem Land zu entwickeln. Mutationen in LQTS kausalen Genen könnten Variabilität in der klinischen Penetranz verleihen, in einem Extrem könnten einige Träger vermutlich völlig gesund sein, aber einige Träger könnten ihre erste Manifestation der Krankheit als Synkope oder plötzlichen Tod haben. Der plötzliche Tod eines kleinen Kindes oder Erwachsenen stellt eine große psychische und emotionale Belastung für die Familie dar, ein Screening der Träger ist unerlässlich, da es einfache Medikamente gibt, z. B. β-Blocker (auch Verhaltensmodifikation), die die Träger sehr effektiv vor den fatalen Folgen von Arrhythmien und SCDs schützen könnten. Personen mit homozygoten Mutationen in den LQTS-Kausalgenen könnten eine schwere Morbidität und extrem hohe Mortalitätsrate aufweisen, einschließlich fetaler Brady-Tachyarrhythmien und in vielen Fällen Fehlgeburten, spontane Abtreibungen bei schwangeren Müttern (12-14).
In Saudi-Arabien wurde auch nicht viel über die Prävalenz verschiedener SNPs in den mit Arrhythmien verbundenen Genen und auch in den Genen, die sie regulieren, geforscht. Splawski et al. (112) beschrieben eine häufige S1103Y-Variante im SCN5A-Gen, die mit Arrhythmie bei Afroamerikanern assoziiert ist. Das als Y1103 bezeichnete Variantenallel ist für die Beschleunigung der Kanalaktivierung verantwortlich, wodurch die Wahrscheinlichkeit von Herzrhythmusstörungen bei Personen afrikanischer Abstammung erhöht wird (52, 112). K393N ist eine Variante des KCNQ1-Gens, die bei LQTS1-Patienten in den USA berichtet wurde, aber in der arabischen Bevölkerung haben wir diese Variante bei 2% der Personen nachgewiesen (unveröffentlichte Daten). Ob die K393N-Variante im KCNQ1-Gen bei Arabern mit der S1103Y (SCN5A) -Variante bei Afroamerikanern oder der D85N (KCNE1) -Variante in der japanischen Bevölkerung vergleichbar ist, könnte ebenfalls untersucht werden (59).
Interessenkonflikterklärung
Die Autoren erklären, dass die Forschung in Abwesenheit von kommerziellen oder finanziellen Beziehungen durchgeführt wurde, die als potenzieller Interessenkonflikt ausgelegt werden könnten.
Danksagung
Wir danken Prof. Arthur Wilde, Prof. Connie Bezzina und dem Herausgeber der Zeitschrift “Heart” für die Erlaubnis, die Abbildung 1 in diesem Manuskript von Ref. (113).
1. Bhuiyan ZA. Klinisches und genetisches Spektrum erblicher Herzrhythmusstörungen. Amsterdam: Universität Amsterdam (2009).
2. Es sind keine frei zugänglichen ergänzenden Materialien verfügbar in: Priori SG, Aliot E, Blømstrom-Lundqvist C, Bossaert L, Breithardt G, Brugada P, et al. Task Force on sudden cardiac death, Europäische Gesellschaft für Kardiologie. Europace (2002) 4:3-18. doi: 10.1053/eupc.2001.0214
Querverweis Volltext
3. Wang Q., Shen J., Splawski I., Atkinson D., Li Z., Robinson JL, et al. SCN5A-Mutationen im Zusammenhang mit einer vererbten Herzrhythmusstörung, Long-QT-Syndrom. Zelle (1995) 80: 805-11. doi:10.1016/0092-8674(95)90359-3
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext
4. Curran MICH, Splawski ich, Timothy KW, Vincent GM, Grün ED, Keating MT. Eine molekulare Grundlage für Herzrhythmusstörungen: HERG-Mutationen verursachen ein Long-QT-Syndrom. Zelle (1995) 80: 795-803. doi:10.1016/0092-8674(95)90358-5
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext
5. Wang Q, Curran ME, Splawski I, Brennen TC, Millholland JM, VanRaay TJ, et al. Positionsklonierung eines neuartigen Kaliumkanalgens: KVLQT1-Mutationen verursachen Herzrhythmusstörungen. Nat Genet (1996) 12:17-23. doi:10.1038/ng0196-17
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext
6. Splawski ich, Tristani-Firouzi M, Lehmann MH, Sanguinetti MC, Keating MT. Mutationen im hminK-Gen verursachen ein Long-QT-Syndrom und unterdrücken die IKs-Funktion. Nat Genet (1997) 17:338-40. doi:10.1038/ng1197-338
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext
7. Sanguinetti MC, Jiang C, Curran MICH, Keating MT. Eine mechanistische Verbindung zwischen einer vererbten und einer erworbenen Herzrhythmusstörung: HERG kodiert den IKr-Kaliumkanal. Zelle (1995) 81: 299-307. doi:10.1016/0092-8674(95)90340-2
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext
8. Neyroud N, Tesson F, Denjoy I, Leibovici M, Donger C, Barhanin J, et al. Eine neuartige Mutation im Kaliumkanalgen KVLQT1 verursacht das kardioauditorische Syndrom von Jervell und Lange-Nielsen. Nat Genet (1997) 15:186-9. doi:10.1038/ng0297-186
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext
9. Kucera JP, Rohr S, Rudy Y. Die Lokalisierung von Natriumkanälen in interkalierten Scheiben moduliert die Herzleitung. Circ Res (2002) 91:1176-82. doi:10.1161/01.RES.0000046237.54156.0A
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext
10. Oxford EM, Musa H, Maass K, Coombs W, Taffet SM, Delmar M. Connexin43 Umbau durch Hemmung der Plakophilin-2-Expression in Herzzellen verursacht. Circ Res (2007) 101:703-11. doi:10.1161/CIRCRESAHA.107.154252
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext
11. Rohr S. Molekulares Übersprechen zwischen mechanischen und elektrischen Übergängen an der interkalierten Scheibe. Circ Res (2007) 101:637-9. doi:10.1161/CIRCRESAHA.107.161901
Querverweis Volltext
12. Bhuiyan ZA, Momenah TS, Gong Q, Amin AS, Ghamdi SA, Carvalho JS, et al. Rezidivierender intrauteriner fetaler Verlust aufgrund nahezu fehlender HERG: klinische und funktionelle Charakterisierung einer homozygoten Nonsense-HERG-Q1070X-Mutation. Herzrhythmus (2008) 5: 553-61. Ursprungsbezeichnung:10.1016/j.hrthm.2008.01.020
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext
13. Bhuiyan ZA, Momenah TS, Amin TS, Al-Khadra AS, Alders M, Wilde AAM, et al. Eine intronische Mutation, die zu einem unvollständigen Überspringen von Exon-2 in KCNQ1 führt, rettet das Gehör beim Jervell- und Lange-Nielsen-Syndrom. Prog Biophysiker Biol (2008) 98:319-27. doi:10.1016/j.pbiomolbio.2008.10.004
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext
14. Bhuiyan ZA, Al-Shahrani S, Al-Khadra AS, Al-Ghamdi S, Al-Kalaf K, Mannens MMAM, et al. Klinische und genetische Analyse des Long-QT-Syndroms bei Kindern aus sechs Familien in Saudi-Arabien: Unterscheiden sie sich? Pediatr Cardiol (2009) 30:490-501. ust-idnr.:10.1007/s00246-008-9377- y
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext
15. Es sind keine frei zugänglichen ergänzenden Materialien verfügbar Zitationen von Shinwari ZM, Al-Hazzani A, Dzimiri N, Tulbah S, Mallawi Y, Al-Fayyadh M, et al. Identifizierung einer neuartigen KCNQ1-Mutation in einer großen saudischen Familie mit Long-QT-Syndrom: klinische Konsequenzen und präventive Implikationen. Clin Genet (2013) 83:370-4. Ursprungsbezeichnung:10.1111/j.1399-0004.2012.01914.x
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext
16. Schwartz PJ, Stramba-Badiale M, Crotti L, Pedrazzini M, Besana A, Bosi G, et al. Prävalenz des angeborenen Long-QT-Syndroms. Auflage (2009) 120: 1761-7. doi:10.1161/CIRCULATIONAHA.109.863209
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext
17. Schwartz PJ, Moss AJ, Vincent GM, Crampton RS. Diagnostische Kriterien für das Long-QT-Syndrom. Update. Auflage (1993) 88: 782-4. doi:10.1161/01.CIR.88.2.782
Querverweis Volltext
18. Cho Y, Behr ER, Berul C, et al. Zusammenfassung: HRS / EHRA / APHRS-Konsenserklärung zur Diagnose und Behandlung von Patienten mit vererbten primären Arrhythmie-Syndromen. Europace (2013) 15:1389-406. doi:10.1093/europace/eut272
Querverweis Volltext
19. Liu JF, Jons C, Moss AJ, McNitt S, Peterson DR, Qi M, et al. Syndrom-Registrierung. Risikofaktoren für rezidivierende Synkopen und nachfolgende tödliche oder fast tödliche Ereignisse bei Kindern und Jugendlichen mit Long-QT-Syndrom. J Am Coll Cardiol (2011) 57:941-50. doi:10.1016/j.jacc.2010.10.025
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext
20. Splawski I, Shen J, Timothy KW, Lehmann MH, Priori S, Robinson JL, et al. Spektrum von Mutationen in Genen des Long-QT-Syndroms. KVLQT1, HERG, SCN5A, KCNE1 und KCNE2. Auflage (2000) 102: 1178-85. doi:10.1161/01.CIR.102.10.1178
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext
21. Westenskow P, Splawski I, Timothy KW, Keating MT, Sanguinetti MC. Zusammengesetzte Mutationen: eine häufige Ursache für ein schweres Long-QT-Syndrom. Auflage (2004) 109: 1834-41. doi:10.1161/01.CIR.0000125524.34234.13
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext
22. Mullally J, Goldenberg I, Moss AJ, Lopes CM, Ackerman MJ, Zareba W, et al. Risiko lebensbedrohlicher kardialer Ereignisse bei Patienten mit langem QT-Syndrom und multiplen Mutationen. Herzrhythmus (2013) 10: 378-82. Ursprungsbezeichnung:10.1016/j.hrthm.2012.11.006
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext
23. Napolitano C, Priori SG, Schwartz PJ, Bloise R, Ronchetti E, Nastoli J, et al. Gentests beim Long-QT-Syndrom: Entwicklung und Validierung eines effizienten Ansatzes zur Genotypisierung in der klinischen Praxis. JAMA (2005) 294:2975-80. doi:10.1001/jama.294.23.2975
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext
24. Roden DM. Klinische Praxis. Long-QT-Syndrom. N Engl J Med (2008) 358:169-76. doi:10.1056/NEJMcp0706513
Querverweis Volltext
25. Schwartz PJ, Priori SG, Spazzolini C, Moss AJ, Vincent GM, Napolitano C, et al. Genotyp-Phänotyp-Korrelation beim Long-QT-Syndrom: Genspezifische Auslöser für lebensbedrohliche Arrhythmien. Auflage (2001) 103: 89-95. doi:10.1161/01.CIR.103.1.89
Querverweis Volltext
26. Ackerman MJ, Tester DJ, Porter CJ. Schwimmen, ein genspezifischer arrhythmogener Auslöser für das vererbte Long-QT-Syndrom. Mayo Clin Proc (1999) 74:1088-94. doi:10.4065/74.11.1088
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext
27. Kapa S, Tester DJ, Salisbury BA, Harris-Kerr C, Pungliya MS, Alders M, et al. Gentests für das Long-QT-Syndrom: Unterscheidung pathogener Mutationen von gutartigen Varianten. Auflage (2009) 120: 1752-60. doi:10.1161/CIRCULATIONAHA.109.863076
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext
28. Moss AJ, Shimizu W, Wilde AA, Towbin JA, Zareba W, Robinson JL, et al. Klinische Aspekte des Typ-1-Long-QT-Syndroms nach Lokalisation, Kodierungstyp und biophysikalischer Funktion von Mutationen, an denen das KCNQ1-Gen beteiligt ist. Auflage (2007) 115: 2481-9. doi:10.1161/CIRCULATIONAHA.106.665406
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext
29. Amin AS, Giudicessi JR., Tijsen AJ, Spanjaart AM, Reckman YJ, Klemens CA, et al. Varianten in der 3′-untranslatierten Region des KCNQ1-kodierten Kv7.1-Kaliumkanals modifizieren den Schweregrad der Erkrankung bei Patienten mit Typ-1-Long-QT-Syndrom allelspezifisch. Eur Herz J (2012) 33:714-23. doi:10.1093/eurheartj/ehr473
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext
30. Barscheshet A, Goldenberg I, O-Uchi J, Moss AJ, Jons C, Shimizu W, et al. Mutationen in zytoplasmatischen Schleifen des KCNQ1-Kanals und das Risiko lebensbedrohlicher Ereignisse: auswirkungen auf das mutationsspezifische Ansprechen auf die β-Blocker-Therapie beim Typ-1-Long-QT-Syndrom. Auflage (2012) 125: 1988-96. doi:10.1161/CIRCULATIONAHA.111.048041
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext
31. Schwartz PJ, Spazolini C, Crotti L, Bathen J, Amlie JP, Timothy K, et al. Das Jervell- und Lange-Nielsen-Syndrom: Naturgeschichte, molekulare Basis und klinisches Ergebnis. Auflage (2006) 113: 783-90. doi:10.1161/CIRCULATIONAHA.105.592899
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext
32. Bhuiyan ZA, Wilde AA. IKs in Herz und Gehör, kann das Ohr mit weniger als das Herz zu tun. Circ Cardiovasc Genet (2013) 6:141-3. doi:10.1161/CIRCGENETICS.113.000143
Querverweis Volltext
33. Jentsch TJ, Wollnik B, Kubisch C, Wieacker P, Esperer HD. Pathophysiologische Mechanismen dominanter und rezessiver KVLQT1-K + -Kanalmutationen bei vererbten Herzrhythmusstörungen. Hum Mol Genet (1997) 6:1943-9. doi:10.1093/hmg/6.11.1943
Querverweis Volltext
34. Doevendans PA, Düren DR, Hauer RN, van Langen IM, Wilde AA, Jongbloed RJ, et al. Hörreize als Auslöser für arrhythmische Ereignisse unterscheiden HERG-bezogene (LQTS2) Patienten von KVLQT1-bezogenen Patienten (LQTS1). J Am Coll Cardiol (1999) 33:327-32. ust-idnr.:10.1016/S0735-1097(98)00578-6
CrossRef Volltext
35. Moss AJ, Zareba W, Kaufman ES, Gartman E, Peterson DR, Benhorin J, et al. Erhöhtes Risiko für arrhythmische Ereignisse beim Long-QT-Syndrom mit Mutationen in der Porenregion des menschlichen Ether-a-go-Go-verwandten Genkaliumkanals. Auflage (2002) 105: 794-9. doi:10.1161/hc0702.105124
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext
36. Lupoglazoff JM, Denjoy I, Bösewicht E, Fressart V, Simon F, Bozio A, et al. Long-QT-Syndrom bei Neugeborenen: Leitungsstörungen im Zusammenhang mit HERG-Mutationen und Sinusbradykardie mit KCNQ1-Mutationen. J Am Coll Cardiol (2004) 43:826-30. doi:10.1016/j.jacc.2003.09.049
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext
37. Es sind keine frei zugänglichen ergänzenden Materialien verfügbar in: Chevalier P, Bellocq C, Millat G, Piqueras E, Potet F, Schott JJ, et al. Torsades de pointes, die den atrioventrikulären Block komplizieren: Hinweise auf eine genetische Veranlagung. Herzrhythmus (2007) 4: 170-4. Ursprungsbezeichnung:10.1016/j.hrthm.2006.10.004
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext
38. Hoorntje T, Alders M, van Tintelen P, van der Lippe K, Sreeram N, van der Wal A, et al. Homozygote vorzeitige Trunkierung des HERG-Proteins: der menschliche HERG-Knockout. Auflage (1999) 100: 1264-7. doi:10.1161/01.CIR.100.12.1264
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext
39. Piippo K, Laitinen P, Schwan H, Toivonen L, Viitasalo M, Pasternack M, et al. Homozygotie für eine HERG-Kaliumkanalmutation verursacht eine schwere Form des Long-QT-Syndroms: Identifizierung einer offensichtlichen Gründermutation bei den Finnen. J Am Coll Cardiol (2000) 35:1919-25. ust-idnr.:10.1016/S0735-1097(00)00636-7
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext
40. Johnson WH Jr, Yang P, Yang T, Lau YR, Mostella BA, Wolff DJ, et al. Klinische, genetische und biophysikalische Charakterisierung einer homozygoten HERG-Mutation, die ein schweres neonatales Long-QT-Syndrom verursacht. Pediatr Res (2003) 53:744-8. doi:10.1203/01.PDR.0000059750.17002.B6
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext
41. Ackerman MJ, Tester DJ, Jones GS, Wird ML, Burrow CR, Curran MICH. Ethnische Unterschiede bei kardialen Kaliumkanalvarianten: auswirkungen auf die genetische Anfälligkeit für plötzlichen Herztod und Gentests auf angeborenes Long-QT-Syndrom. Mayo Clin Proc (2003) 78:1479-87. doi:10.4065/78.12.1479
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext
42. Es sind keine frei zugänglichen ergänzenden Materialien verfügbar Zitation Crotti L, Lundquist AL, Pedrazzini M, Ferrandi C, De Ferrari GM, et al. KCNH2-K897T ist ein genetischer Modifikator des latenten angeborenen Long-QT-Syndroms. Auflage (2005) 112: 1251-8. doi:10.1161/CIRCULATIONAHA.105.549071
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext
43. Nof E, Cordeiro JM, Pérez GJ, Scornik FS, Calloe K, Liebe B, et al. Ein häufiger Einzelnukleotidpolymorphismus kann das Long-QT-Typ-2-Syndrom verschlimmern, was zu einem plötzlichen Kindstod führt. Circ Cardiovasc Genet (2010) 3:199-206. doi:10.1161/CIRCGENETICS.109.898569
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext
44. Bezzina C, Veldkamp MW, van Den Berg MP, Postma AV, Rook MB, Viersma JW, et al. Eine einzelne Na (+) -Kanalmutation, die sowohl Long-QT- als auch Brugada-Syndrome verursacht. Circ Res (1999) 85:1206-13. doi:10.1161/01.RES.85.12.1206
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext
45. Kyndt F., Probst V., Potet F., Demolombe S., Chevallier J.C., Baro I., et al. Neuartige SCN5A-Mutation, die entweder zu einem isolierten Herzleitungsdefekt oder zum Brugada-Syndrom in einer großen französischen Familie führt. Auflage (2001) 104: 3081-6. doi:10.1161/hc5001.100834
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext
46. Es sind keine frei zugänglichen ergänzenden Materialien verfügbar in: Makita N, Behr E, Shimizu W, Horie M, Sunami A, Crotti L, et al. Die E1784K-Mutation in SCN5A ist mit einem gemischten klinischen Phänotyp des Typ-3-Long-QT-Syndroms assoziiert. J Clin Invest (2008) 118:2219-29. doi:10.1172/JCI34057
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext
47. Keller DI, Acharfi S, Delacrétaz E, Benammar N, Rotter M, Pfammatter JP, et al. Eine neuartige Mutation in SCN5A, delQKP 1507-1509, verursacht langes QT-Syndrom: Rolle des Q1507-Rückstands bei der Natriumkanalinaktivierung. J Mol Cell Cardiol (2003) 35:1513-21. doi:10.1016.2003.08.007
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext
48. Es sind keine frei zugänglichen ergänzenden Materialien verfügbar Zitationen Priori SG, Schwartz PJ, Napolitano C, Ronchetti E, Grillo M, et al. Risikostratifizierung beim Long-QT-Syndrom. N Engl J Med (2003) 348:1866-74. doi:10.1056/NEJMoa022147
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext
49. Lupoglazoff JM, Cheav T, Baroudi G, Berthet M, Denjoy I, Cauchemez B, et al. Homozygote SCN5A-Mutation beim Long-QT-Syndrom mit funktioneller Zwei-zu-Eins-atrioventrikulärer Blockade. Circ Res (2001) 89:E16–21. doi:10.1161/hh1401.095087
Querverweis Volltext
50. Zhang Y, Yang C, Huang C, Zhou X, Qiang H, et al. Die kardiale Natriumkanalmutation delQKP 1507-1509 ist assoziiert mit dem expandierenden phänotypischen Spektrum von LQT3, Leitungsstörung, dilatative Kardiomyopathie und hohe Inzidenz von plötzlichem Jugendtod. Europace (2008) 10:1329-35. doi:10.1093/europace/eun202
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext
51. Zumhagen S, Veldkamp MW, Stallmeyer B, Baartscheer A, Eckardt L, Paul M, et al. Eine heterozygote Deletionsmutation im kardialen Natriumkanalgen SCN5A mit Verlust- und Funktionsgewinnmerkmalen manifestiert sich als isolierte Leitungskrankheit ohne Anzeichen eines Brugada- oder Long-QT-Syndroms. PLoS One (2013) 8:e67963. doi:10.1371/Zeitschrift.pone.0067963
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext
52. Anlage LD, Bowers PN, Liu Q, Morgan T, Zhang T, Zustand MW, et al. Eine häufige kardiale Natriumkanalvariante im Zusammenhang mit dem plötzlichen Kindstod bei Afroamerikanern, SCN5A S1103Y. J Clin Invest (2006) 116: 430-5. doi:10.1172/JCI25618
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext
53. Mohler PJ, Schott JJ, Gramolini AO, Dilly KW, Guatimosim S, duBell WH, et al. Die Ankyrin-B-Mutation verursacht Typ-4-Long-QT-Herzrhythmusstörungen und plötzlichen Herztod. Natur (2003) 421: 634-9. doi:10.1038/nature01335
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext
54. Schott JJ, Charpentier F, Peltier S, Foley P, Drouin E, Bouhour JB, et al. Kartierung eines Gens für das Long-QT-Syndrom auf Chromosom 4q25-27. Bin J Hum Genet (1995) 57:1114-22.
Pubmed Zusammenfassung / Pubmed Volltext
55. Barhanin J, Lesage F, Guillemare E, Fink M, Lazdunski M, Romey G. K (V) LQT1- und lsK (minK) -Proteine assoziieren sich zur Bildung des I (Ks) -Herzkaliumstroms. Natur (1996) 384: 78-80. doi:10.1038/384078a0
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext
56. Sanguinetti MC, Curran ME, Zou A, Shen J, Spector PS, Atkinson DL, et al. Koassemblierung von K (V) LQT1- und I (IsK) -Proteinen zur Bildung des kardialen I (Ks) -Kaliumkanals. Natur (1996) 384: 80-3. doi:10.1038/384080a0
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext
57. Schulze-Bahr E, Wang Q, Wedekind H, Haverkamp W, Chen Q, Sun Y, et al. KCNE1-Mutationen verursachen das Jervell- und Lange-Nielsen-Syndrom. Nat Genet (1997) 17:267-8. doi:10.1038/ng1197-267
Querverweis Volltext
58. Duggal P, Vesely MR, Wattanasirichaigoon D, Villafane J, Kaushik V, Beggs AH. Mutation des Gens für IKs, das sowohl mit Jervell- als auch mit Lange-Nielsen- und Romano-Ward-Formen des Long-QT-Syndroms assoziiert ist. Auflage (1998) 97: 142-6. doi:10.1186/1471-2350-9-24
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext
59. Nishio Y, Makiyama T, Itoh H, Sakaguchi T, Ohno S, Gong YZ, et al. D85N, ein KCNE1-Polymorphismus, ist eine krankheitsverursachende Genvariante beim Long-QT-Syndrom. J Am Coll Cardiol (2009) 54:812-9. doi:10.1016/j.jacc.2009.06.005
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext
60. Paulussen AD, Gilissen RA, Armstrong M, Doevendans PA, Verhasselt P, Smeets HJ, et al. Genetische Variationen von KCNQ1, KCNH2, SCN5A, KCNE1 und KCNE2 bei arzneimittelinduzierten Patienten mit langem QT-Syndrom. Mol Med (2004) 82:182-8. ust-idnr.:10.1007/s00109-003-0522- z
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext
61. Abbott GW, Sesti F, Splawski I, Buck ME, Lehmann MH, Timothy KW, et al. MiRP1 bildet mit HERG IKr-Kaliumkanäle und ist mit Herzrhythmusstörungen assoziiert. Zelle (1999) 97: 175-87. doi:10.1016/S0092-8674(00)80728-X
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext
62. Es sind keine frei zugänglichen ergänzenden Materialien verfügbar in: Gordon E, Panaghie G, Deng L, Bee KJ, Roepke TK, Krogh-Madsen T, et al. Eine KCNE2-Mutation bei einem Patienten mit Herzrhythmusstörungen, die durch Hörreize und ein Ungleichgewicht der Serumelektrolyte hervorgerufen wird. Cardiovasc Res (2008) 77:98-106. doi:10.1093/cvr/cvm030
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext
63. Tawil R, Tristani-Firouzi M, Canún S, Bendahhou S, Tsunoda A, et al. Mutationen in Kir2.1 verursachen die entwicklungs- und episodischen elektrischen Phänotypen des Andersen-Syndroms. Zelle (2001) 105: 511-9. doi:10.1016/S0092-8674(01)00342-7
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext
64. Kubo Y, Baldwin TJ, Jan YN, Jan LY. Primärstruktur und funktionelle Expression eines Maus-internen Kaliumkanals. Natur (1993) 362: 127-33. doi:10.1038/362127a0
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext
65. Raab-Graham KF, Radeke CM, Vandenberg CA. Molekulare Klonierung und Expression eines menschlichen Herzens über einen Kaliumkanal. Neuroreport (1994) 5: 2501-5. doi:10.1097/00001756-199412000-00024
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext
66. Decher N, Kumar P, Bloise R, Splawski I, Timothy KW, Sharpe LM, et al. Ca (V) 1.2 Kalziumkanaldysfunktion verursacht eine Multisystemstörung einschließlich Arrhythmie und Autismus. Zelle (2004) 119: 19-31. doi:10.1016/j.Zelle.2004.09.011
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext
67. Splawski I, Timothy KW, Decher N, Kumar P, Sachse FB, Beggs AH, et al. Schwere Arrhythmie-Störung, die durch kardiale L-Typ-Kalziumkanalmutationen verursacht wird. Proc Natl Acad Sci USA (2005) 102:8089-96. doi:10.1073/pnas.0502506102
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext
68. Gillis J, Buraschnikow E, Antzelevitch C, Blaser S, Gross G, Turner L, et al. Langes QT, Syndaktylie, Gelenkkontrakturen, Schlaganfall und neuartige CACNA1C-Mutation: Erweiterung des Spektrums des Timothy-Syndroms. Bin J Mit Genet EIN (2012) 158EIN:182-7. doi:10.1002/ajmg.a.34355
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext
69. Dufendach KA, Giudicessi JR., Boczek NJ, Ackerman MJ. Mütterlicher Mosaikismus verwirrt die neonatale Diagnose des Typ-1-Timotheus-Syndroms. Pädiatrie (2013) 131: e1991–5. doi:10.1542/peds.2012-2941
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext
70. Vatta M, Ackerman MJ, Ye B, Makielski JC, Ughanze EE, Taylor EW, et al. Mutant Caveolin-3 induziert persistierenden späten Natriumstrom und ist mit Long-QT-Syndrom assoziiert. Auflage (2006) 114:2104–12. doi:10.1161/CIRCULATIONAHA.106.635268
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext
71. Cronk LB, Ye B, Kaku T, Tester DJ, Vatta M, Makielski JC, et al. Neuartiger Mechanismus für den plötzlichen Kindstod: persistierender später Natriumstrom infolge von Mutationen in Caveolin-3. Herzrhythmus (2007) 4: 161-6. Ursprungsbezeichnung:10.1016/j.hrthm.2006.11.030
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext
72. Engelman JA, Zhang X, Galbiati F, Volonte D, Sotgia F, Pestell RG, et al. Molekulare Genetik der Caveolin-Genfamilie: Auswirkungen auf menschliche Krebsarten, Diabetes, Alzheimer-Krankheit und Muskeldystrophie. Bin J Hum Genet (1998) 63:1578-87. doi:10.1086/302172
CrossRef Volltext
73. Balijepalli RC, Foell JD, Halle DD, Hölle JW, Kamp TJ. Die Lokalisierung von Ca (2 +) -Kanälen vom L-Typ des Herzens zu einem caveolären makromolekularen Signalkomplex ist für die beta (2) -adrenerge Regulation erforderlich. Proc Natl Acad Sci USA (2006) 103:7500-5. doi:10.1073/pnas.0503465103
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext
74. Medeiros-Domingo A, Kaku T, Tester DJ, Iturralde-Torres P, Itty A, Ye B, et al. SCN4B-kodierte Natriumkanal-Beta4-Untereinheit beim kongenitalen Long-QT-Syndrom. Auflage (2007) 116: 134-42. doi:10.1161/CIRCULATIONAHA.106.659086
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext
75. MJ, Marquardt ML, Tester DJ, Ackerman MJ, Sampson KJ, Kass RS. Die Mutation eines A-Kinase-Ankerproteins verursacht ein Long-QT-Syndrom. Proc Natl Acad Sci USA (2007) 104:20990-5. doi:10.1073/pnas.0710527105
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext
76. Wu G., Ai T., Kim JJ, Mohapatra B., Xi Y., Li Z., und andere. Alpha-1-Syntrophin-Mutation und das Long-QT-Syndrom: eine Erkrankung der Natriumkanalstörung. Circ Arrhythm Electrophysiol (2008) 1:193-201. doi:10.1161/CIRCEP.108.769224
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext
77. Yang Y, Yang Y, Liang B, Liu J, Li J, Grunnet M, et al. Identifizierung eines Kir3.4 mutation im angeborenen Long-QT-Syndrom. Bin J Hum Genet (2010) 86:872-80. Ursprungsbezeichnung:10.1016/j.ajhg.2010.04.017
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext
78. Roden DM. Mechanismen und Management von Proarrhythmie. Am J Cardiol (1998) 82:49I–57I. doi:10.1016/S0002-9149(98)00472-X
Querverweis Volltext
79. Mitcheson JS, Chen J, Lin M, Culberson C, Sanguinetti MC. Eine strukturelle Grundlage für das arzneimittelinduzierte Long-QT-Syndrom. Proc Natl Acad Sci USA (2000) 97:12329-33. doi:10.1073/pnas.210244497
Querverweis Volltext
80. Kannankeril PJ, Roden DM. Arzneimittelinduzierte lange QT und Torsade de Pointes: jüngste Fortschritte. Curr Opin Cardiol (2007) 22:39-43. doi:10.1097/HCO.0b013e32801129eb
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext
81. Veerman CC, Verkerk AO, Blom MT, Klemens CA, Langendijk PN, van Ginneken AC, et al. Langsam verzögerte Gleichrichter-Kaliumstromblockade trägt wesentlich zum arzneimittelinduzierten Long-QT-Syndrom bei. Circ Arrhythm Elektrophysiol (2013) 6:1002-9. doi:10.1161/CIRCEP.113.000239
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext
82. Sesti F, Abbott GW, Wei J, Murray KT, Saksena S, Schwartz PJ, et al. Ein häufiger Polymorphismus im Zusammenhang mit Antibiotika-induzierten Herzrhythmusstörungen. Proc Natl Acad Sci USA (2000) 97:10613-8. doi:10.1073/pnas.180223197
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext
83. Kääb S, Crawford DC, Sinner MF, Behr ER, Kannankeril PJ, Wilde AA, et al. Eine große Kandidatengenstudie identifiziert den KCNE1 D85N-Polymorphismus als möglichen Modulator von arzneimittelinduzierten Torsades de pointes. Circ Cardiovasc Genet (2012) 5:91-9. doi:10.1161/CIRCGENETICS.111.960930
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext
84. Nakamura K, Katayama Y, Kusano KF, Haraoka K, Tani Y, Nagase S, et al. Anti-KCNH2-Antikörper-induziertes Long-QT-Syndrom: neuartige erworbene Form des Long-QT-Syndroms. J Am Coll Cardiol (2007) 50:1808-9. doi:10.1016/j.jacc.2007.07.037
Querverweis Volltext
85. Moss AJ, Zareba W, Benhorin J, Locati EH, Halle WJ, Robinson JL, et al. EKG-T-Wellenmuster in genetisch unterschiedlichen Formen des erblichen Long-QT-Syndroms. Auflage (1995) 92: 2929-34. doi:10.1161/01.CIR.92.10.2929
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext
86. Zhang L, Timothy KW, Vincent GM, Lehmann MH, Fuchs J, Giuli LC, et al. Spektrum von ST-T-Wellenmustern und Repolarisationsparametern beim kongenitalen Long-QT-Syndrom: EKG-Befunde identifizieren Genotypen. Auflage (2000) 102: 2849-55. doi:10.1161/01.CIR.102.23.2849
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext
87. Schulze-Bahr E., Schulze-Bahr T., Schulze-Bahr E., Schulze-Bahr E., Schulze-Bahr E., Schulze-Bahr E., Schulze-Bahr E., et al. Genotypspezifischer Beginn von Arrhythmien beim kongenitalen Long-QT-Syndrom: mögliche Therapieimplikationen. Auflage (2006) 114: 2096-103. doi:10.1161/CIRCULATIONAHA.106.642694
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext
88. In: Locati EH, Zareba W, Moss AJ, Schwartz PJ, Vincent GM, Lehmann MH, et al. Alters- und geschlechtsspezifische Unterschiede in den klinischen Manifestationen bei Patienten mit angeborenem Long-QT-Syndrom: Befunde aus dem Internationalen LQTS-Register. Auflage (1998) 97: 2237-44. doi:10.1161/01.CIR.97.22.2237
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext
89. Zareba W, Moss AJ, Schwartz PJ, Vincent GM, Robinson JL, Priori SG, et al. Einfluss des Genotyps auf den klinischen Verlauf des Long-QT-Syndroms. Internationale Forschungsgruppe für das Long-QT-Syndrom-Register. N Engl J Med (1998) 339:960-5. doi:10.1056/NEJM199810013391404
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext
90. Goldenberg I, Moss AJ, Bradley J, Polonsky S, Peterson DR, McNitt S, et al. Long-QT-Syndrom nach dem 40. Auflage (2008) 117:2192-201. doi:10.1161/CIRCULATIONAHA.107.729368
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext
91. Zareba W, Moss AJ, Locati EH, Lehmann MH, Peterson DR, Halle WJ, et al. Syndrom-Registrierung. Modulierende Auswirkungen von Alter und Geschlecht auf den klinischen Verlauf des Long-QT-Syndroms nach Genotyp. J Am Coll Cardiol (2003) 42:103-9. ust-idnr.:10.1016/S0735-1097(03)00554-0
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext
92. Seth R, Moss AJ, McNitt S, Zareba W, Andrews ML, Qi M, et al. Long-QT-Syndrom und Schwangerschaft. J Am Coll Cardiol (2007) 49:1092-8. doi:10.1016/j.jacc.2006.09.054
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext
93. Schwartz PJ, Priori SG, Dumaine R, Napolitano C, Antzelevitch C, Stramba-Badiale M, et al. Eine molekulare Verbindung zwischen dem plötzlichen Kindstod und dem Long-QT-Syndrom. N Engl J Med (2000) 343:262-7. doi:10.1056/NEJM200007273430405
Querverweis Volltext
94. Schwartz PJ, Priori SG, Bloise R, Napolitano C, Ronchetti E, Piccinini A, et al. Molekulare Diagnose bei einem Kind mit plötzlichem Kindstod. Lanzette (2001) 358: 1342-3. ust-IDNR.:10.1016/S0140-6736(01)06450-9
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext
95. Christiansen M, Tønder N, Larsen LA, Andersen PS, Simonsen H, Oyen N, et al. Mutationen im HERG K + -Ionenkanal: eine neuartige Verbindung zwischen dem Long-QT-Syndrom und dem plötzlichen Kindstod. Am J Cardiol (2005) 95:433-4. Ursprungsbezeichnung:10.1016/j.amjcard.2004.09.054
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext
96. Tester DJ, Ackerman MJ. Postmortales Long-QT-Syndrom Gentests für plötzlichen unerklärlichen Tod bei jungen Menschen. J Am Coll Cardiol (2007) 49:240-6. doi:10.1016/j.jacc.2006.10.010
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext
97. Hofman N, Tan HL, Clur SA, Alders M, van Langen IM, Wilde AA. Beitrag der erblichen Herzkrankheit zum plötzlichen Herztod in der Kindheit. Pädiatrie (2007) 120: e967–73. doi:10.1542/peds.2006-3751
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext
98. Wedekind H, Schulze-Bahr E, Arnold R, Veldkamp MW, Bajanowski T, et al. De-novo-Mutation im SCN5A-Gen im Zusammenhang mit dem frühen Einsetzen des plötzlichen Kindstodes. Auflage (2001) 104: 1158-64. doi:10.1161/hc3501.095361
Querverweis Volltext
99. Wang DW, Desai RR, Crotti L, Arnestad M, Insolia R, Pedrazzini M, et al. Kardiale Natriumkanalfunktionsstörung beim plötzlichen Kindstod. Auflage (2007) 115: 368-76. doi:10.1161/CIRCULATIONAHA.106.646513
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext
100. Van Norstrand DW, Valdivia CR, Tester DJ, Ueda K, London B, Makielski JC, et al. Molekulare und funktionelle Charakterisierung neuartiger Glycerin-3-phosphat-Dehydrogenase-1-ähnlicher Genmutationen (GPD1-L) beim plötzlichen Kindstod. Auflage (2007) 116: 2253-9. doi:10.1161/CIRCULATIONAHA.107.704627
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext
101. Tan BH, Pundi KN, Van Norstrand DW, Valdivia CR, Tester DJ, Medeiros-Domingo A, et al. Plötzlicher Kindstod-assoziierte Mutationen in den Beta-Untereinheiten des Natriumkanals. Herzrhythmus (2010) 7: 771-8. Ursprungsbezeichnung:10.1016/j.hrthm.2010.01.032
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext
102. Miller TE, Estrella E, Myerburg RJ, Garcia de Viera J, Moreno N, Rusconi P, et al. Rezidivierender Verlust des Fötus im dritten Trimester und mütterlicher Mosaikismus für das Long-QT-Syndrom. Auflage (2004) 109:3029-34. doi:10.1161/01.CIR.0000130666.81539.9E
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext
103. Es sind keine frei zugänglichen ergänzenden Materialien verfügbar in: Chockalingam P, Crotti L, Girardengo G, Johnson JN, Harris KM, van der Heijden JF, et al. Nicht alle Betablocker sind bei der Behandlung des Long-QT-Syndroms Typ 1 und 2 gleich: höhere Rezidive von Ereignissen unter Metoprolol. J Am Coll Cardiol (2012) 60:2092-6. doi:10.1016/j.jacc.2012.07.046
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext
104. Schwartz PJ, Priori SG, Cerrone M, Spazzolini C, Odero A, Napolitano C, et al. Sympathische Denervation des linken Herzens bei der Behandlung von Hochrisikopatienten, die vom Long-QT-Syndrom betroffen sind. Auflage (2004) 109: 1826-33. doi:10.1161/01.CIR.0000125523.14403.1E
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext
105. Singh B, al Shahwan SA, Habbab MA, al Deeb SM, Biary N. Idiopathisches Long-QT-Syndrom: Die richtige Frage stellen. Lanzette (1993) 341:741. doi:10.1016/0140-6736(93)90501-7
CrossRef Volltext
106. Gorgels AP, Al Fadley F, Zaman L, Kantoch MJ, Al Halees Z. Das Long-QT-Syndrom mit gestörter atrioventrikulärer Überleitung: eine maligne Variante bei Säuglingen. J Cardiovasc Elektrophysiol (1998) 9:1225-32. doi:10.1111/j.1540-8167.1998.tb00096.x
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext
107. Kantoch MJ, Qurashi MM, Bulbul ZR, Gorgels AP. Ein Neugeborenes mit einer komplexen angeborenen Herzkrankheit, einer atrioventrikulären Blockade und einer ventrikulären Torsade de Pointes-Tachykardie. Pacing Clin Elektrophysiol (1998) 21: 2664-7. doi:10.1111/j.1540-8159.1998.tb00043.x
Querverweis Volltext
108. El-Hazmi MA, al-Swailem AR, Warsy AS, al-Swailem AM, Sulaimani R, al-Meshari AA. Blutsverwandtschaft unter der saudi-arabischen Bevölkerung. J Med Genet (1995) 32:623-6. Ursprungsbezeichnung: 10.1136/jmg.32.8.623
Querverweis Volltext
109. El Mouzan MI, Al Salloum AA, Al Herbish AS, Qurachi MM, Al Omar AA. Blutsverwandtschaft und schwere genetische Störungen bei saudischen Kindern: eine gemeindebasierte Querschnittsstudie. Ann. Med (2008) 28:169-73. doi:10.4103/0256-4947.51726
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext
110. Medeiros-Domingo A, Bhuiyan ZA, Tester DJ, Hofman N, Bikker H, van Tintelen JP, et al. Der RYR2-kodierte Ryanodin-Rezeptor / Calcium-Freisetzungskanal bei Patienten, bei denen zuvor entweder eine katecholaminerge polymorphe ventrikuläre Tachykardie oder ein Genotyp-negatives, belastungsinduziertes Long-QT-Syndrom diagnostiziert wurde: eine umfassende offene Leserahmen Mutationsanalyse. J Am Coll Cardiol (2009) 54:2065-74. doi:10.1016/j.jacc.2009.08.022
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext
111. Al-Aama JY, Al-Ghamdi S, Bdier AY, Wilde AA, Bhuiyan ZA. De novo-Mutation im KCNQ1-Gen, die zum Jervell- und Lange-Nielsen-Syndrom führt. Clin Genet (2013). Ursprungsbezeichnung:10.1111/cge.12300
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext
112. Splawski I, Timothy KW, Tateyama M, Clancy CE, Malhotra A, Beggs AH, et al. Variante des SCN5A-Natriumkanals mit Risiko für Herzrhythmusstörungen. Wissenschaft (2002) 297: 1333-6. doi:10.1126/Wissenschaft.1073569
Querverweis Volltext
113. Wilde AA, Bezzina CR. Genetik von Herzrhythmusstörungen. Herz (2005) 91: 1352-8.