A genetika ajak-és szájpadhasadék
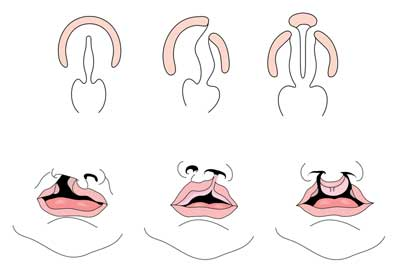 Intro/abstractCleft ajak szájpadhasadékkal vagy anélkül egy komplex veleszületett anomália, amely izolálható vagy más rendellenességekkel együtt látható. Része lehet a genetikai szindróma fenotípusának is. Ez a cikk az ajak-és szájpadhasadék prevalenciájának, a kiújulás kockázatainak és az egyéb veleszületett rendellenességek kockázatainak áttekintésére szolgál. Genetikai szindrómák és teratogén expozíciók, amelyekről ismert, hogy orális hasadékokkal járnak, feltárják. Ezenkívül a gyermekgyógyászati klinikai genetikai környezetben általában kért genetikai tesztek az ajak-és szájpadhasadékkal rendelkező beteg értékeléséről lesz szó.
Intro/abstractCleft ajak szájpadhasadékkal vagy anélkül egy komplex veleszületett anomália, amely izolálható vagy más rendellenességekkel együtt látható. Része lehet a genetikai szindróma fenotípusának is. Ez a cikk az ajak-és szájpadhasadék prevalenciájának, a kiújulás kockázatainak és az egyéb veleszületett rendellenességek kockázatainak áttekintésére szolgál. Genetikai szindrómák és teratogén expozíciók, amelyekről ismert, hogy orális hasadékokkal járnak, feltárják. Ezenkívül a gyermekgyógyászati klinikai genetikai környezetben általában kért genetikai tesztek az ajak-és szájpadhasadékkal rendelkező beteg értékeléséről lesz szó.
szájpadhasadékkal vagy anélkül (CL/CP) különbözik az izolált szájpadhasadéktól (CP) embrionális, epidemiológiai és genetikai szinten. Az ajakhasadék jellemzően a maxilláris kiemelkedés és a mediális orr kiemelkedés eredménye, amely nem olvad össze az embrionális fejlődés ötödik és hatodik hete között. A normális szájpadlás az elsődleges és a másodlagos szájpad kialakulásából származik. Az elsődleges szájpad a hat-hét héten alakul ki a mediális orr -, laterális orr-és maxilláris folyamatok kialakulásával és fúziójával. A másodlagos szájpad a palatális polcokból származik (amelyek az első elágazási ív páros maxilláris folyamataiból fejlődnek ki) vízszintessé válnak és összeolvadnak, így az embrionális fejlődés kilencedik hetében kialakulnak a kemény és lágy szájpadok. A polcok az elsődleges szájpadlással és az orrszeptummal is összeolvadnak. (1)
az orális hasadékok az újszülöttek egyik leggyakoribb születési rendellenessége Óvoda, Általános prevalenciája 1.6 ezer újszülöttre világszerte, a CL/CP körülbelül ezer születésnél, a CP pedig 0,6 ezer születésnél. (2) a CL/CP gyakorisága magasabb az ázsiai, afrikai és indián származású egyéneknél. A CL / CP férfiaknál is gyakoribb. Ezzel szemben nincs szignifikáns különbség a CP előfordulásában a különböző etnikai hátterek között, a CP pedig gyakoribb a nőknél. (3) A családon belüli kiújulás kockázata attól függ, hogy a hasadék izolált-e (más klinikai leletek nélkül), vagy genetikai szindróma részének tekintik-e. Az orális hasadékok legtöbb esetét izolálják (körülbelül 80%). Az izolált hasadékokról úgy gondolják, hogy multifaktoriális öröklődéssel rendelkeznek: több tényező kombinációjának köszönhetők, mind genetikai, mind környezeti tényezők. A kiújulás kockázata (1.táblázat) növekszik, ha egynél több érintett rokon van. Az ismétlődés kockázata is növekszik, annál súlyosabb a hiba.

ajak-és szájpadhasadék látható más veleszületett rendellenességek. A genetikai vagy teratogén etiológia valószínűsége növeli a veleszületett anomáliákat, amelyekkel a beteg bemutatja. Más kérdések, például értelmi fogyatékosság, viselkedési problémák, például autizmus, dysmorphic jellemzők vagy más orvosi problémák jelenléte szintén valószínűbbé teszi a genetikai rendellenességet vagy a teratogén expozíciót. Az ajakhasadékkal rendelkező egyének körülbelül 13% – ának lesz más orvosi problémája vagy rendellenessége. Ez a szám 37% – ra nő az ajak-és szájpadhasadék esetén, és 47% – ra csak a szájpadhasadék esetén.
a teratogén szerek (például talidomid, antikonvulzív szerek, alkohol, retinsav és cigaretta) és az anyai betegségek (például cukorbetegség, rubeola és folsavhiány) prenatális expozíciója bizonyítottan növeli az orális hasadékok kockázatát. Az amniotikus sávok jelenléte szintén növeli a hasadékok kockázatát. A perikonceptuális folsav-kiegészítésről ismert, hogy csökkenti az orális hasadékok kockázatát.
a Pierre Robin szekvencia egy craniofacialis anomália, melyet mandibularis hypoplasia vagy micrognathia, másodlagos U-alakú szájpadhasadék és glossoptosis jellemez, ami obstruktív apnoéhoz és táplálkozási nehézségekhez vezet. A Pierre Robin szekvencia a genetikai szindrómák részének tekinthető (22q11.2 deléciós szindróma, Stickler-szindróma; az alábbiakban ismertetjük). (5)
az orális hasadékokkal kapcsolatos genetikai szindrómák százai vannak, beleértve a citogenetikai rendellenességeket (aneuploidiák, mikrodeléciók) és az egygénes (Mendeli) rendellenességeket. A genetikai diagnózis megerősítése elengedhetetlen a prognózis meghatározásához és a kiújulás kockázatának megállapításához.
az olyan Aneuploidiák, mint a 13-as és 18-as triszómia, szoros kapcsolatban állnak a CL/CP-vel. A 13-as triszómia (más néven Patau-szindróma) a 13.kromoszóma három példányával vagy a 13. kromoszómát érintő kiegyensúlyozatlan Robertson-transzlokációkkal társul. Az ilyen állapotban született csecsemők általában az újszülöttkorban halnak meg. A klinikai jellemzők közé tartozik az ajak – és szájpadhasadék, növekedési retardáció, súlyos központi idegrendszeri rendellenességek (beleértve a holoprosencephaly-t is), mikrocefália, micropthalmia, iris coloboma, a szemek hiánya, rosszul formált fülek, polydactyly, összeszorított ököllel, rocker alsó lábakkal, veleszületett szívhibák és urogenitális hibák. A középvonali hasadékok (egyébként nagyon ritkák) a 13.triszómiában megfigyelhetők a középvonal hibáinak kockázata miatt, beleértve a holoprosencephaly-t is. A 18-as triszómiát (más néven Edwards-szindrómát) általában a 18-as kromoszóma három különálló példánya okozza, és a szülés utáni rossz kimenetelhez kapcsolódik. A klinikai jellemzők közé tartozik az ajak-és szájpadhasadék, az értelmi fogyatékosság, a gyarapodás elmulasztása, a veleszületett szívbetegség, a hypertonia, a micrognathia, a rövid szegycsont, az alacsonyan beállított hibás fülek, az összeszorított kezek, a rocker alsó lábak és a hipoplasztikus körmök. A 13-as és 18-as triszómia könnyen igazolható vagy kizárható kromoszóma-elemzéssel (kariotipizálás).
a Mikrodeletiós szindrómák általában a kromoszóma egy részének delécióját jelentik. Ezek a deléciók túl kicsik lehetnek ahhoz, hogy standard kariotipizálással kimutathatók legyenek, és FISH (fluoreszcencia in situ hibridizáció) vagy microarray technológia kimutatására lehet szükség. A szájpadhasadékhoz kapcsolódó jól ismert mikrodeletion szindróma a 22q11.2 deléciós szindróma (más néven Digeorge/Velocardiofacialis szindróma). Palatális rendellenességek, beleértve a velopharyngealis inkompetenciát, a submucosalis hasadékokat, a bifid uvulát és a szájpadhasadékot az egyének 69% – ánál észlelték 22q11.2 delécióval, és a Pierre Robin szekvencia része lehet. Egyéb klinikai eredmények közé tartozik a veleszületett szívbetegség, halláskárosodás, dysmorphic jellemzők, immunhiány, hypocalcaemia, vese anomáliák, táplálkozási problémák, csontváz anomáliák és pszichiátriai rendellenességek. Úgy gondolják, hogy a 22q11.2 deléciós szindróma eseteinek körülbelül 10% – a családi. A deléció autoszomális domináns módon szegregálódik.(6) A Wolf-Hirschhorn-szindróma, amely a 4.kromoszóma rövid karjában történő deléciónak köszönhető, szintén orális hasadékokkal jár (az érintett egyének 25-50% – ában). Jellegzetes arcvonások (beleértve a kiemelkedő glabellát, amely a “görög harcos sisak megjelenéséhez” vezet), veleszületett szívbetegség, értelmi fogyatékosság, rohamok, gyarapodás elmulasztása, micrognathia, preauricularis címkék vagy gödrök, valamint hypodontia szintén az állapot részének tekinthetők.(7)
az orális hasadékokkal járó Egygénes rendellenességek közé tartozik a Stickler-szindróma, a Treacher Collins-szindróma és a Van der Woude-szindróma. A Stickler-szindróma egy autoszomális domináns kollagén rendellenesség, ritkábban autoszomális recesszív öröklődés. Közös jellemzők: szájpadhasadék (Pierre Robin szekvencia részeként vagy mikrognathia nélkül), halláskárosodás (szenzorineurális és vezetőképes), csontváz leletek (korai kezdetű ízületi gyulladás, spondyloepiphysealis dysplasia), szem anomáliák (magas myopia, üvegtesti rendellenességek) és jellegzetes arcvonások (a maxilla és az orrhíd fejletlenségével, a középső retrúzióval). A Stickler-szindróma genetikai vizsgálata összetett lehet, mivel az érintett egyénekben legalább hat gén mutációját írták le. A Stickler-szindrómában szenvedő betegek körülbelül 90% – ánál mutációk vannak a COL2A1 génben, és az állapot autoszomális domináns formája van.(8) A Treacher Collins-szindróma egy autoszomális domináns állapot, amelyet szájpadhasadék jellemez ajakhasadékkal vagy anélkül az érintett egyének 28% – ában. Egyéb rendellenességek közé tartozik a járomcsontok és az állkapocs hypoplasia, a külső fül anomáliái, az alsó szemhéj coloboma, vezetőképes halláskárosodás, az alsó szempillák hiánya, a preauricularis haj elmozdulása az arcra, valamint a choanalis stenosis vagy atresia. A Treacher Collins-szindróma diagnózisa klinikai és radiológiai eredményeken alapul. Legalább három gén mutációit írták le, a TCOF1 mutációit a betegek 78-93% – ánál figyelték meg.(9) A Van der Woude-szindrómát veleszületett, általában bilaterális, paramedián alsó ajakfistulák (gödrök), vagy néha kis halmok jelenléte jellemzi, amelyek sinus traktussal vezetnek az ajak nyálkahártyájából, valamint orális hasadékok (beleértve a CL/CP-t és a CP-t). Van der Woude egy autoszomális domináns állapot, amely az IRF6 gén mutációihoz kapcsolódik (10). Az egygénes vagy többgénes állapotok vizsgálata a gén közvetlen elemzését igényli szekvenálással és/vagy deléciós / duplikációs elemzéssel (például MLPA).
tekintettel arra, hogy az ajak-és szájpadhasadékkal járó genetikai szindrómák összekapcsolhatók aneuploidiákkal, kromoszóma mikrodeléciókkal/mikroduplikációkkal vagy egygénes rendellenességekkel, a genetikai tesztelés bonyolult folyamat lehet. Az alapos kórtörténet, a három generációs törzskönyv, a terhességi előzmények és a klinikai genetikus dysmorphology vizsgálata tisztázhatja a klinikai képet, és lehetővé teszi a célzott genetikai vizsgálatokat. Az újabb technológiák, beleértve a microarray-t, lehetővé teszik a kis mikrodeléciók és mikroduplikációk azonosítását, amelyeket korábban a szokásos kariotipizálás elmulasztott. Sajnos ez a technika az ismeretlen klinikai jelentőségű deléciók és duplikációk azonosításához is vezet, ami bonyolítja a genetikai tanácsadási folyamatot. Az egygénes rendellenességek vagy a mendeli rendellenességek vizsgálata megköveteli a kívánt gén genetikai vizsgálatának klinikai elérhetőségét. Drága is lehet, ha nem fedezi az egészségügyi biztosítás. Az új technológiák, például a következő generációs szekvenálás, az exome szekvenálás vagy a genom szekvenálás (együttesen genomikai tesztek néven ismert) klinikailag elérhetővé váltak. Több száz-ezer gén egyidejű elemzésével ezek a tesztek jelentősen növelik a diagnosztikai teljesítményt és a hozamot. Más technikákkal összehasonlítva ezek a tesztek gyorsabban és költséghatékonyabban adhatnak választ. A kutatási területen az exome és a genom szekvenálása új gének azonosításához, valamint a genetikai mutációk klinikai jellemzőinek és spektrumának bővítéséhez vezetett. A microarray technológiához hasonlóan a genomikai tesztek olyan szindrómákat is kimutathatnak, amelyek nem kapcsolódnak a beteg megjelenéséhez és/vagy a tesztelés okához. Tekintettel a genetikai tesztelés eredendő összetettségére, tájékozott beleegyezés szükséges.
következtetés
bár az ajak-és szájpadhasadék az esetek többségében elszigetelt anomália, szoros kapcsolat van az orális hasadékok és más anomáliák és genetikai szindrómák között. A klinikai genetikus és a genetikai tanácsadó által végzett genetikai értékelés elengedhetetlen a megelőző útmutatáshoz és a kiújulás kockázatának meghatározásához. A genetikai vizsgálat, amely tájékozott beleegyezést igényel, koordinálható és értelmezhető a genetikai értékelés során.
anya Revah, MS, A New York-i Brooklynban található Maimonides csecsemők és Gyermekkórház Orvosi Genetikai osztályának vezető genetikai tanácsadója. Aktív tagja a Maimonides Medical Center és a Kings County Hospital Cleft Lip and Palate multidiszciplináris csapatának. A bostoni Bostoni Egyetemen genetikai tanácsadásból szerzett mesterképzést.
1. Sadler TW. Langman orvosi Embriológiája. Kilencedik Kiadás. 390-395. oldal.
2. Parker SE, Mai CT, Canfield MA, Rickard R, Wang Y, Meyer RE, Anderson P, Mason CA, Collins JS, Kirby RS, Correa A. A Nemzeti születési rendellenességek megelőzési hálózat számára. Frissített nemzeti születési prevalencia becslések a kiválasztott születési rendellenességekről az Egyesült Államokban. 2004-2006. Születési rendellenességek kutatása (A. rész): klinikai és molekuláris Teratológia 2010;88:1008-1016.
3. Fraser FC. Az ajak-és szájpadhasadék genetikája. Az vagyok. J. Hum. Genet. 1970;22: 336–352.
4. Van Rooij IA, Ocke MC, et al. A perikonceptuális folátbevitel kiegészítéssel és táplálékfelvételgel csökkenti a nem szindrómás ajakhasadás kockázatát szájpadhasadékkal vagy anélkül. Előző Med 2004; 39: 689-694.
5. Tan TY. Kilpatrick N, Farlie PG. A Pierre Robin szekvencia fejlődési és genetikai perspektívái. Az vagyok. J. Med. Genet. 2013;163C: 295-305.
6. McDonald-McGinn DM, Emanuel BS, Zackai EH. 22q11.2 deléciós szindróma. Szeptember. 23, 1999. . Ban ben: Pagon RA, Adam MP, Ardinger HH, et al., szerkesztők. Általános Vélemények . Seattle (WA): Washingtoni Egyetem, Seattle; 1993-2014. Elérhető: http://www.ncbi.nlm.nih.gov/books/NBK1523/.
7. Battaglia a, Carey JC, Déli utca, et al. Wolf-Hirschhorn Szindróma. Április. 29, 2002. . Ban ben: Pagon RA, Adam MP, Ardinger HH, et al., szerkesztők. Általános Vélemények . Seattle (WA): Washingtoni Egyetem, Seattle; 1993-2014. Elérhető: http://www.ncbi.nlm.nih.gov/books/NBK1183/.
8. Robin NH, Moran RT, Ala-Kokko L. Stickler szindróma. Jun. 9, 2000. . Ban ben: Pagon RA, Adam MP, Ardinger HH, et al., szerkesztők. Általános Vélemények . Seattle (WA): Washingtoni Egyetem, Seattle; 1993-2014. Elérhető: http://www.ncbi.nlm.nih.gov/books/NBK1302/.
9. Katsanis SH, Jabs EW. Treacher Collins Szindróma. Jul. 20, 2004. . Ban ben: Pagon RA, Adam MP, Ardinger HH, et al., szerkesztők. Általános Vélemények . Seattle (WA): Washingtoni Egyetem, Seattle; 1993-2014. Elérhető: http://www.ncbi.nlm.nih.gov/books/NBK1532/.
10. Schutte BC, Saal HM, Goudy S, et al. IRF6-tal kapcsolatos rendellenességek. Október. 30, 2003. . Ban ben: Pagon RA, Adam MP, Ardinger HH, et al., szerkesztők. Általános Vélemények . Seattle (WA): Washingtoni Egyetem, Seattle; 1993-2014. Elérhető: http://www.ncbi.nlm.nih.gov/books/NBK1407/.