Autoszomális recesszív Congenital ichthyosis / Actas Dermo-Sifiliogr adapt
Bevezetés
az ichthyosis legújabb konszenzusos osztályozása 2 fő formát különböztet meg: a nem szindrómás formákat, amelyek csak a bőr megnyilvánulásaival vannak jelen, valamint a szindrómás formákat, amelyek más szervekben is megnyilvánulásokkal vannak jelen (1.táblázat).1 A nem szindrómás formák közül 4 csoportot azonosítanak: közös ichthyoses, autoszomális recesszív veleszületett ichthyoses (ARCIs), keratinopátiás ichthyoses és más kevésbé gyakori ichthyoses.Hagyományosan az ARCIs csoportot 2 rendellenességre, lamelláris ichthyosisra (LI) és veleszületett ichthyosiform erythrodermára (CIE) osztották. Az új osztályozásban harlequin ichthyosis (HI) került ebbe a csoportba1,mert az ABCA12 gén inaktiváló mutációit azonosították felelősnek ezért a rendellenességért,2,3, míg ugyanazon gén nonszensz mutációi LI4 vagy CIE5, 6 fenotípust eredményezhetnek. Az ARCIs csoportba tartozó egyéb kevésbé gyakori változatok az öngyógyító collodion baby (SHCB), az acral SHCB és a fürdőruha ichthyosis.7-9
konszenzus osztályozás az ichthyosis klinikai jellemzői alapján1.
| Nemdromikus formák | szindrómás formák |
| közös IchthyosesIchthyosis vulgarisrecessív x-kapcsolt ichthyosis (nem szindrómás )ajimajor formákharlequin ichthyosislamelláris ichthyosisveleszületett ichthyosiform erythrodermaMinor formáköngyógyító kollódium babaakrális öngyógyító kollódium babafürdőruha ichthyosiskeratinopátiás IchthyosesMajor formákepidermolitikus ichthyosisfelületes epidermolitikus ichthyosisminor formsannularis epidermolytic ichthyosiscurth-Macklin Ichthyosisautosomal recesszív epidermolytic ichthyosisEpidermolytic nevusegyéb Formákloricrin keratodermaErythrokeratodermia vararabilispeeling skin syndromeCongenital reticularis ichthyosiform erythrodermaKLICK szindróma | Syndromic X-linked Ichthyosrecessive x-linked ichthyosis (syndromic)ichthyosis follicularis, alopecia és photophobia (IFAP) szindrómaconradi-Scammann-happle szindróma (chondrodysplasia punctata Típus 2)szindrómás autoszomális ichthyosisskin rendellenességeknetherton Szindrómaichthyosis-hypothrichosis szindrómaichthyosis-sclerosing cholangitis szindrómaichthyosis-sclerosing cholangitis szindrómarichothystrophyneurológiai rendellenességekj xhamgren-Larsson syndromeRefsum diseaseMEDNIK syndromeFatal disease courseGaucher disease, type 2Multiple sulfatase deficiencyCEDNIK syndromeARC syndromeOther associated signsKID syndromeChanarin-Dorfman syndromeIchthyosis prematurity syndrome |
Abbreviations: ARC, arthrogryposis–renal dysfunction–cholestasis; ARCI, autosomal recessive congenital ichthyosis; CEDNIK, cerebral dysgenesis, neuropathy, ichthyosis, and palmoplantar keratoderma; KID, keratitis ichthyosis deafness; KLICK, keratosis linearis with ichthyosis congenital and sclerosing keratoderma; MEDNIK, mentális retardáció, enteropathia, süketség, perifériás neuropathia, ichthyosis, keratoderma.
az ARCIs epidemiológiájáról csak korlátozott adatok állnak rendelkezésre. Az Egyesült Államokban a születéskori prevalencia a LI esetében 1 / 100 000 populáció, a CIE esetében pedig 1 / 200 000 populáció. Más tanulmányok szerint a LI és a CIE együttes prevalenciája 1 / 200 000-300 000 populáció.10,11 egyes országokban, például Norvégiában, a becsült prevalencia nagyobb (1 per 91 000) az alapító mutációk miatt.12 egy populációban 1 vagy több ismétlődő mutáció megállapítása lehet azért, mert a mutáció a történelem egy adott pontján következett be, majd generációról generációra öröklődött (alapító mutáció), vagy azért, mert a genom azon régiójában, ahol a mutációt megtalálják, mutációra érzékeny DNS-szekvencia van (mutációs hotspot). Spanyolországban az ARCI becsült prevalenciája az általános populációban 1/138 000, a 1/61 700 pedig a 10 év alatti gyermekek körében.13 Spanyolország egyes régióiban az előfordulás még magasabb lehet. A galíciai partvidéken például 1/33 000 prevalenciáról számoltak be, ami az alapító hatásnak is köszönhető.14
lamelláris Ichthyosis és veleszületett ichthyosiform ErythrodermaClinical jellemzők
bár eredetileg azt hitték, hogy a LI és a CIE különböző entitások, beszámoltak olyan betegekről, akiknek köztes klinikai tünetei voltak, és mindkét állapotot ugyanazon gén mutációi okozhatják.15,16 ezenkívül az azonos mutációval rendelkező betegek, még ugyanabban a családban is, különböző fenotípusokat alakíthatnak ki.12,15
a legtöbb beteg kollódium membránba burkolva születik, amely az élet első heteiben fokozatosan eltűnik, és helyébe a végleges fenotípus lép (ábra. 1A). Hypohidrosis, súlyos hő intolerancia és körömdisztrófia gyakran megfigyelhető mind LI, mind CIE-ben.17-19 LI-ben szenvedő betegnél általában súlyosabb klinikai tünetek jelentkeznek, mint a CIE-ben szenvedőknél. Nagy, gyakran sötét színű, lemezszerű pikkelyeik vannak, amelyek az egész testfelületet lefedik. Az eritroderma hiányzik vagy minimális. Az ilyen betegeknél általában ectropion és időnként eclabium, ízületi és orrporc hypoplasia, hegesedés alopecia, különösen a fejbőr szélén, és palmoplantaris keratoderma (ábra. 1B és C). A CIE-t az eritroderma jelenléte és a finom fehéres méretezés jellemzi (ábra. 2). Néhány betegnél erythema és generalizált méretezés jelentkezett. A pikkelyek nagyok és sötét színűek lehetnek, különösen a lábak extenzor felületén. Kevésbé súlyos esetekben az erythema enyhe, a méretezés pedig rendben van.

a lamelláris ichthyosis klinikai jellemzői. A, barnás lamellás hámlás. B, jelzett talpi hyperkeratosis. C, a fejbőr hegesedése.
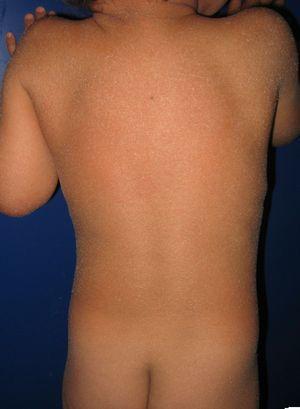
veleszületett ichthyosiform erythrodermában és az ALOXE3 gén mutációiban szenvedő beteg. Enyhe erythema és generalizált fehéres furfuraceous hámlás látható.
hisztopatológia
a hisztopatológiai elváltozások nem adnak diagnózist. LI-ben masszív ortokeratotikus hyperkeratosis figyelhető meg, általában kétszer olyan kiterjesztéssel, mint a CIE-ben. Az epidermisz akantotikus, esetenként pikkelysömörszerű megjelenést mutat. A sejtproliferációs sebesség normális vagy enyhén emelkedett.17-19 Cie-ben szenvedő betegnél kevésbé markáns hyperkeratosis, fokális vagy kiterjedt parakeratosis, normál vagy megvastagodott szemcsés réteg, kifejezettebb acanthosis. Az epidermális forgalom növekszik.17-19
ULTRASTRUKTÚRA
bár a molekuláris, klinikai és ultrastrukturális eredmények közötti szoros összefüggést eddig nem találtak, az elektronmikroszkópia mindazonáltal hasznos lehet az ichthyosis egyéb formáinak kizárására és egyes esetekben a genetikai elemzések irányítására. A veleszületett ichthyosis négy típusát írták le (2.táblázat).
a veleszületett Ichtiózisok ultrastrukturális osztályozása.
| Típus | fő jellemző | egyéb jellemzők | mutációk | klinikai tünetek |
| 1 | a 2., 3. és 4. típusú ichthyosis ultrastrukturális markereinek hiánya | lipidcseppek vagy gyűrűk a stratum corneumban (Leggyakoribb)kis keratohyalin granulesvesicularis vagy lobularis membrán bevonat granulátumok | TGM1 (33.3%)ALOX12B (2 eset) | CIE |
| 2 | koleszterin hasadékok a stratum corneumban | korhadt burkolatok hiánya vagy elvékonyodásakis keratohyalin granuláklipid cseppek | TGM1 (89-100%) | LI |
| 3 | laminált membrán szerkezetek a stratum granulosumban és / vagy a stratum corneumban. | abnormális membrán bevonat granulesLipid cseppekfoci kiemelkedő juxtanuclear vakuolák a szemcsés réteg | NIPAL4 (93%) | CIE (leggyakoribb)LI |
| 4 | Trilamelláris membrán csomagok, amelyek kitöltik a stratum granulosum és/vagy stratum corneum egyes sejtjeit | rendellenes membránbevonat granulátumok | FTAP4 | ichthyosis koraszülöttségi szindróma (100%) |
rövidítések: Cie, veleszületett ichthyosiform erythroderma; LI, lamelláris ichthyosis.
1. típusú veleszületett Ichthyosis
az 1. típusú veleszületett ichthyosisra a 2., 3. és 4. típusú ichthyosis ultrastrukturális markereinek hiánya jellemző. Ezért a diagnózist általában csak akkor végzik el, ha a többi típust kizárták. A leggyakoribb megállapítás a lipidcseppek vagy gyűrűk jelenléte a stratum corneumban (ábra. 3A).20 ezek a lipidcseppek nem állandó jellemzők vagy specifikusak erre a bizonyos típusra,mivel nem minden esetben vannak jelen, 20 és más típusú ichthyosisban is jelen lehetnek.21,22 klinikailag a legtöbb betegnél CIE manifesztáció jelentkezik.12,20 a betegek egyharmadában mutációk vannak a TGM1 génben.16 ezt az ultrastrukturális típust az ALOX12B gén mutációival összefüggésben is azonosították.23,24

elektronmikroszkóp képek. A, 1. típusú veleszületett ichthyosis, amely lipidcseppeket mutat a stratum corneumban, valamint a többi típusú ichthyosis ultrastrukturális markereinek hiányát. B, 2. típusú veleszületett ichthyosis, amelyet a corneocytákban koleszterin-hasadékok (nyíl) jelenléte jellemez.
2. típusú veleszületett Ichthyosis
a 2. típusú veleszületett ichthyosisra a stratum corneum koleszterin-hasadékai jellemzőek (ábra. 3B).21 Az ilyen hasadékok állandó leletek az ilyen típusú ichthyosisban, és ugyanazon beteg különböző biopsziáiban kimutathatók; az orális retinoidokkal végzett kezelés nincs hatással ezekre a hasadékokra.12,25 elektron-sűrű aggregátumot is megfigyeltek a corneocytákon néhány hiányos tgáz-1 aktivitású betegnél.26-28 klinikailag a legtöbb betegnél a CIE súlyos megnyilvánulása van.12 Ez az ultrastrukturális típus szorosan kapcsolódik a tgm1 gén mutációihoz.12,16
3. típusú veleszületett Ichthyosis
a 3.típusú veleszületett ichthyosisra a stratum granulosum és/vagy stratum corneum lamelláris membránszerkezetei jellemzőek. Ezek a szerkezetek csíkokban vannak elrendezve a maghoz közeli üres tér körül.22,29-31 Az ilyen típusú klinikai tünetek különböznek a többitől; az ichthyosis kialakulása változó, a hámlás és az erythema foltos vagy általános lehet, és különösen a hajlításokat érinti. A NIPAL4 gén mutációi felelősek a 3.típusú ichtiózisok 93% – áért.32
4. típusú veleszületett Ichthyosis
jellemző, hogy a 4.típusú veleszületett ichthyosisban a stratum granulosum és a stratum corneum egyes sejtjei trilamelláris membráncsomagokkal vannak feltöltve.33 ezek az eredmények patognomikusak az ichthyosis koraszülöttségi szindróma esetében, amely állapot jelenleg az ichthyosis szindrómás formájának tekinthető.34,35
molekuláris vizsgálatok
genetikai szempontból az Arci-k nagyon heterogének. A legtöbb esetben a TGM1 gén társul, de 5 másik gén (ALOX12B, ALOXE3, NIPAL4, CYP4F22 és ABCA12) mutációit jelentették. Fischer et al.36 vizsgált 520 család ARCI és azonosított mutációk legalább 1 ilyen gének 78% – ában (TGM1 32%, NIPAL4 16%, ALOX12B 12%, CYP4F22 8%, ALOXE3 5% és ABCA12 5%). Egy másik vizsgálatban, amelyben 250, különböző eredetű ARCI-vel rendelkező beteg vett részt, 38% – uk volt TGM1 mutáció, 6,8% – UK volt ALOXE3 mutáció, 6,8% – uk pedig ALOX12B mutáció.37 Galíciában a vizsgált családok 75% – ában tgm1, ALOX12B, ALOXE3, NIPAL4 és CYP4F22 gének mutációit azonosítottuk, de a mutációk megoszlása eltérő volt.14 a TGM1 gént 68-ban mutálták.Az esetek 7%-a, míg az ALOXE3 gént csak 1 betegben mutálták. A többi 3 vizsgált gén egyikében sem találtunk mutációt.
TGM1
a TGM1 gén a 14q11. 2 kromoszómán található, és 15 exonnal rendelkezik (GenBank NM-000359.2). Kódolja a Tgáz 1 enzimet, amely az epidermiszben található 3 Tgáz enzim egyike.38 Ez az enzim részt vesz a korhadt burok kialakulásában azáltal, hogy katalizálja számos fehérje, például az involukrin, a lorikrin és a prolinban gazdag fehérjék kalciumfüggő térhálósodását.39,40 azt is katalizálja kötődése ??- hidroxi-ceramidok a korhadt boríték külső rétegében, a belső rétegben lévő fehérjékkel.41,42 TGM1 mutációban szenvedő betegeknél a korhadt burok hiányzik, a TGase 1 aktivitás csökkent vagy nem létezik.43-47
1995 óta, amikor ezt a gént azonosították az ARCI egyes eseteiért,48-50-nél több mint 110 mutációt jelentettek különböző eredetű betegeknél. A TGM1 mutációi az ARCI leggyakoribb okai.36,37 ezt a mutációt az Egyesült Államokban az esetek 55% – ában, Norvégiában pedig az esetek 84% – ában találták meg.12,51 a leggyakoribb mutáció c.877-2A> G, amelyet az eddig jelentett mutált allélek 34% – ában találtak.52 a mutáció magas gyakorisága olyan országokban, mint az Egyesült Államok és Norvégia, az alapító hatásnak köszönhető.12,53 a második leggyakoribb mutáció P. Arg142His. Ilyen és hasonló mutációkat jelentettek olyan országokban, mint Egyiptom, Németország, Finnország és az Egyesült Államok,15,49-51,54-56, és úgy tűnik, hogy ezek hotspot mutációk.57 A p.Arg307Trp mutáció gyakori a japán populációban.5 Galíciában, a P. Arg760X, c.1223_1227delacaca és c.984 + 1G>a mutációk a TGM1-ben a családok 81,82% – ában azonosítottak mutációkat ebben a génben, ami alapító hatásra utal.14 ennek a hipotézisnek a megerősítését haplotípus-vizsgálat (még nem publikált munka).
a TGM1 mutációk felelősek az LI15 legtöbb esetéért,27,44,46,56,58-63 a CIE esetek kis százalékában.43,47,64,65 az ilyen mutációk az ARCI más formáit is előidézhetik, mint például az SHCB, az acral SHCB és a fürdőruha ichthyosis.
számos tanulmány megkísérelte kimutatni a genotípus-fenotípus összefüggéseket a tgm1 mutációi és az ultrastrukturális vagy klinikai eredmények között, de eddig nem figyeltek meg szignifikáns összefüggést.15,16,53 általában a tgm1 gén mutációival rendelkező betegek súlyosabban érintettek, mint az ilyen mutációk nélküli betegek. Egy 83, Svédországban és Észtországban ARCI-ben szenvedő betegen végzett vizsgálatban az ectropion és a collodion baby jelenlétét TGM1 mutációkkal társították, míg az erythema magasabb arányát figyelték meg azoknál a betegeknél, akiknél nem volt mutáció ebben a génben.66 egy másik tanulmány kimutatta, hogy a tgm1 mutációk hordozói és nem hordozói között a skálázás típusa a fő különbség, mivel megállapította, hogy az ebben a génben mutációval rendelkező betegek lamelláris skálázással rendelkeznek, míg a TGM1 mutációval nem rendelkezők 80% – a finom skálázással rendelkezik.14 ezenkívül megfigyelték, hogy a csonka mutációk gyakrabban társulnak hypohidrosissal és izzadási rendellenességekkel, mint a missense mutációk.51 az észak-amerikai populációban egy bizonyos klinikai jellemzők jelenlétén alapuló modell azt jósolja, hogy azok a betegek, akik kollodion csecsemőként születtek, és szemészeti rendellenességekkel és/vagy alopeciával rendelkeznek, 4-szer nagyobb valószínűséggel rendelkeznek TGM1 mutációkkal.51
ALOXE3 és ALOX12B
az ALOXE3 és ALOX12B gének a 17p13.1.67 kromoszómán helyezkednek el, hasonló szerkezetűek, 15 exonnal, amelyek az epidermális LOXs eLOX-3 és 12R-LOX kódolását végzik.68,69 az a tény, hogy túlnyomórészt az epidermisz szuprabasalis rétegeiben fejeződnek ki, támogatja szerepüket az epidermális differenciálódás előrehaladott fázisaiban, a lamellás testek feldolgozásában való részvétellel.24,70 ezek az enzimek a hepoxilin útvonal szomszédos lépéseire hatnak (ábra. 4). A 12R-LOX az arachidonsavat 12R-hidroxi-eikozát-tetraénsavvá alakítja,míg az eLOX-3 Ezt a terméket epoxi-alkohol izomerré alakítja69, 71 a hepoxilin A3 család.72 a hepoxilin készítmény instabil, és a sejtekben egy specifikus trihidroxi-származékká (trioxilin) hidrolizálódik. Bár a hepoxilin útvonal termékeinek pontos szerepe nem ismert, feltételezték, hogy részt vehetnek a stratum corneum intercelluláris lipidjeinek kialakulásában, vagy jelekként szolgálhatnak a keratinocita differenciálódás kiváltására.
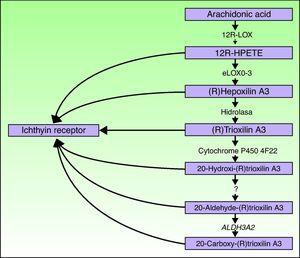
a hepoxilin útvonal vázlatos diagramja, amely bemutatja az ALOXE3, ALOX12B, NIPAL4 és CYP4F22 gének részvételét. Ezeknek a géneknek a mutációi felelősek az ARCI bizonyos típusaiért. A HPETE hidroperoxi-eikozát-tetraénsavat jelez.
az ALOX12B és ALOXE3 géneket először 2002-ben azonosították.73,74 azóta több mint 30 mutációt jelentettek az ALOX12B génben23, 24,37,75-77 és körülbelül 10 az ALOXE3 génben37,74,75. Ezek a mutációk felelősek az Arcis 14-17% – áért36, 37 és 72.2% SHCBs.23,78,79 a mutációk és a fenotípus közötti ok-okozati összefüggést igazolták annak bizonyításával, hogy az epidermális LOX katalitikus aktivitása teljesen megszűnt az ilyen mutációkkal rendelkező betegekben75, 80, valamint olyan állatmodellek alkalmazásával, amelyek reprodukálják az emberben látható ichthyosiform fenotípust.81-83 mindkét gén felelős az ARCI esetek hasonló százalékáért. Az ALOXE3 gén különböző mutációinak tartománya azonban korlátozott, mivel 2 mutáció, a p.Arg234X és a P.Pro630Leu dominál, amelyek látszólag megfelelnek a hotspotoknak.37,74,75
az ALOXE3 és ALOX12B gének mutációival rendelkező betegek általában CIE fenotípust mutatnak.74,75,77 a méretezés súlyossága enyhe vagy közepes, a pikkelyek fehéres vagy világosbarna színűek. Erythema is jelen lehet. A betegek 76% – a kollodion csecsemőként születik, 88% – UK izzadási rendellenességekkel rendelkezik.37 Az ALOX12B gén mutációival rendelkező betegek korlátozottabb, fehéres hámlást mutatnak, mint az ALOXE3 gén mutációinak hordozói. Ezekben az esetekben a mérleg barnás és tapadó. Az erythema, a palmoplantáris hyperkeratosis jelenléte, valamint a palmoplantáris redők hangsúlyozása szintén összefügg az ALOX12B mutációkkal.37
Ichthyin/NIPAL4
a NIPAL4 gén, más néven ichthyin gén, az 5q33 kromoszómán található. 6 exonja van, amelyek több, ismeretlen funkciójú transzmembrán doménnel rendelkező fehérjét kódolnak.84 feltételezték, hogy a fehérjetermék ugyanabban a metabolikus útvonalban vesz részt, mint a LOX, és receptorként működhet az A3 és B3 trioxilinek vagy a hepoxilin metabolikus útvonalának más metabolitjai számára.84 így szerepet játszik a lamellás testek kialakulásában vagy az extracelluláris tér felé történő szállításukban.32 ennek alátámasztására 2 észrevétel. Először is, az esetek 93% – ában a gén mutációi a 3.típusú veleszületett ichthyosis ultrastrukturális mintázatához kapcsolódnak, amelyet a lamelláris testek rendellenességei és a hosszúkás perinukleáris membránok jelenléte jellemez a stratum granulosumban.32 másodperc alatt a NIPAL4 lényegében az epidermisz granulosum rétegében expresszálódik, ahol a lamelláris testek jelen vannak.85
a NIPAL4 gén 2004-es felfedezése óta 84 csak 9 mutációt jelentettek mediterrán országokból (Algéria, Törökország és Szíria), 84 skandináv országból,32 Pakisztánból,85 Feröer szigetről,32-ből és Dél-Amerikából.84
a gén mutációival rendelkező betegek klinikai spektruma széles, még ugyanazon család tagjai között is. 3,7% 32-60% 84 kollódium csecsemőként születik. Amikor a kollódium membrán eltűnik, a legtöbb betegnél a CIE megnyilvánulása alakul ki, finom fehéres pikkelyekkel az arcon és a törzsön erythemás alapon, és nagyobb, barnás pikkelyekkel a nyakon, a fenéken és a lábakon.84 markáns xerosis, generalizált barnás retikuláris hiperkeratotikus plakkok, amelyek a bőr redőiben hangsúlyosnak tűnnek, és az arc diszkrómiája jelen lehet.32,85 ezenkívül a palmoplantaris keratoderma gyakori lelet, alkalmi ujjkontraktúrákkal és ívelt ujjkörmökkel együtt. Egyes tanulmányok az LI-re jellemzőbb eredményekről számoltak be.32,85 néhány betegnél az atópiás dermatitis jeleinek és tüneteinek jelenlétéről számoltak be, bár az FLG gén mutációit ezen esetek egyikében sem mutatták ki.85
CYP4F22
az FLJ39501 vagy CYP4F22 gén a 19p13.12.86 kromoszómán található, 12 exonja van87 és egy P450 citokrómot kódol, 4.család, F alcsalád, 2. polipeptid, a leukotrién B4 – homogén-hidroxiláz (CYP4F2). Az FLJ39501 terméke által a bőrben és a reakció szubsztrátjaiban katalizált reakció a cyp4f2 és CYP4F3 ismert homológjaival analóg módon levezethető.88 feltételezték, hogy a cyp4f2 és a CYP4F3 részt vesz a hepoxilin útvonalban azáltal, hogy katalizálja a trioxilin A3 átalakulását 20-hidroxi-(R)trioxilin A387-re, és hogy ennek az útvonalnak a végterméke, a 20-karboxi-trioxilin A3 kulcsfontosságú biológiai szabályozó hatást fejthet ki a bőrön.89
a mai napig ennek a génnek csak 8 mutációját jelentették 12 mediterrán országokból87 és 1 izraeli származású családban.62
a családok által jelentett Leaf Enterprises et al.,87 a betegek többségének születésekor CIE fenotípusa volt, ez később LI-re progrediált. a betegek általában kifejezett erythrodermával születtek, bár kollódiummembrán nélkül. Ahogy öregedtek, általánosított fehéres-szürke pikkelyeket fejlesztettek ki, amelyek a periumbilicalis régióban, a fenéken és a test alsó részén voltak markánsabbak. Gyakori volt a tenyér és a talp hiperlinearitása, valamint a fejbőr hámlása, a pityriasiform típus idején.87 egy másik családban az érintett 3 tag collidion csecsemőként született, és intenzív erythroderma, generalizált desquamation és palmoplantaris keratoderma alakult ki.62
ABCA12
2003-ban beszámoltak arról, hogy az ABCA12 gén felelős a LI egyes eseteiért, és a 2q34.4 kromoszómához térképezték fel.A 2, 3abca12 53 exont kódol, és az ABC transzporterek családjába tartozik, amelyek megkötik az adenozin-trifoszfátot, miközben elősegítik több molekula szállítását a sejtmembránon keresztül.90 az ABCA alcsalád tagjai mind részt vesznek a lipid transzportban.91 a hiányos ABCA12 funkció lipidtranszport rendellenességeket okoz a lamellás testekben, így az intercelluláris lipidszint csökkenéséhez vezet a stratum corneumban.3az infrastrukturális vizsgálatok kimutatták, hogy az ABCA12 a glikozil-ceramidokhoz kapcsolódó lamelláris testekben található.A 91abca12 mutációk összefüggésbe hozhatók a glikozil-ceramidok eloszlásának és transzportjának rendellenességeivel, valamint az intercelluláris térben a lipid gát egyik fő összetevőjének, a hidroxiceramidoknak a csökkenésével.3,6,92,93 az ezeknél a betegeknél előforduló masszív hyperkeratosis kompenzáló válasz lehet a hiányos lipid gátra.94 ennek oka lehet A corneocyták hámlásának hiánya is, 93 amelyet bizonyos proteázok, például a kallikrein 5 és a katepszin D transzportjának a lamelláris testek rendellenességeiből eredő hibái okozhatnak.95 Egérmodellek és in vitro vizsgálatok arra utalnak, hogy az ABCA12 mutációk hatással vannak az epidermális differenciálódásra is.95-97
a mai napig több mint 50 mutációt jelentettek az ABCA12 génben Afrikából, Európából, Pakisztánból és Japánból származó ARCI-ben szenvedő betegeknél. A leggyakoribb mutációk P.Val244serfster28,2,98,99 azonosított Pakisztáni és indiai populációk, és p.Asn1380Ser,4 azonosított afrikai családok. Mindkét esetben ezek alapító mutációk lehetnek.
az ABCA12 mutációk mértéke a fenotípushoz kapcsolódik, a funkció teljes elvesztésével járó mutációk pedig a HI fenotípushoz vezetnek.2,3,98 – 102 ezzel szemben LI és CIE esetében a legtöbb mutáció missense, és kevésbé súlyos hatással van a fehérje működésére.4-6, 103 úgy tűnik, hogy az LI fenotípus alapjául szolgáló mutációk az első adenozin-trifoszfátot kötő kazetta régióban koncentrálódnak.4 klinikailag a CIE-ben szenvedő betegek és az ABCA12 gén mutációi közepes méretű skálákkal rendelkeznek, amelyek valamivel nagyobbak, mint az ilyen fenotípusú betegeknél általában megfigyeltek.
Harlequin Ichthyosis
a HI vagy a harlequin fetus az ichthyosis súlyos és általában halálos formája. A gyermekek általában koraszülöttek, kiterjedt, fényes hiperkeratotikus plakkokkal, mély repedésekkel elválasztva, amelyek az egész bőrt lefedik, és geometriai mintákat képeznek, amelyek a Harlekin által viselt ruházatra emlékeztetnek, ezáltal az állapot nevét adják. A bőr feszessége a szemhéjak és az ajkak jelentős elfordulásához, az ízületi és orrporc kezdetleges fejlődéséhez, és esetenként mikrocefáliához vezet. A gyermekeknek ritkán van szempillájuk vagy szemöldökük, bár a fejbőrön lévő haj konzerválódhat. A kezek és a lábak duzzadtak és ödémásak, és gyakran kesztyűszerű réteg borítja őket. Lehet, hogy ujj kontraktúrájuk van.
az ilyen betegeknél az újszülöttkori halálozás kockázata nagyon magas.104 a tüdőszellőztetés sérül; a transzepidermális vízveszteség kiszáradáshoz, hidroelektromos egyensúlyhiányhoz és termikus instabilitáshoz vezet; és a fertőzések kockázata megnő. Az arc szorítása és az eclabium gátolja a szopást és ezért az etetést,ennek megfelelően a kiszáradás súlyosbodása. Az ilyen állapotú újszülöttek ritkán éltek hosszabb ideig néhány hétig. Az utóbbi években azonban a hosszú távú túlélés esélye jelentősen megnőtt, elsősorban a szisztémás retinoidok beadása és az intenzív újszülöttgondozás előrehaladása miatt.105 egy nemrégiben végzett vizsgálatban az orális retinoidokkal kezelt betegek 83% – a túlélte a kezeletlen betegek 24% – át. A legtöbb haláleset az élet első 3 napjában történt, de a kezelést csak ezt követően kezdték meg sok túlélőnél.104 ez azt sugallja, hogy sok ilyen korai haláleset a retinoid kezeléstől függetlenül történt volna.
az újszülött időszakot túlélő gyermekeknél általában súlyos CIE alakul ki.106 az abca12 gén mutációinak jellege és helye, valamint a transzporter funkció elvesztésének mértéke határozhatja meg a prognózist.3,92,107 azoknak a betegeknek, akik bizonyos fokú fehérje aktivitást tartanak fenn, bár minimálisak, nagyobb esélyük lehet a túlélésre. A homozigóta mutációk hordozói magasabb halálozási arányt mutatnak.104
a HI fő szövettani jellemzője a rendkívül vastag és tömör ortokeratotikus stratum corneum jelenléte. A szőrtüszők és a verejtékcsatornák kiemelkedő hiperkeratotikus dugókkal rendelkeznek107,108 és kóros vagy hiányzó lamelláris testek, lipid zárványok vagy organellák vagy magok maradványai vannak a corneocytákban, és az ultrastrukturális vizsgálatban az intercelluláris lipidek hiánya.108 109 a szőrtüszők jelentős keratotikus anyag koncentrációt mutatnak,ami a Hi diagnosztikai jellemzője a prenatális diagnózishoz.
a mai napig az ABCA12 gén mutációinak kimutatási aránya a HI-ben szenvedő betegeknél közel 100%, tehát ez genetikailag homogén állapotnak tűnik.
Collodion baba és öngyógyító Collodion baba
Collodion babák általában koraszülöttek és perinatális morbiditás és mortalitás nőtt. Születéskor az újszülöttet fényes tanított átlátszó membrán borítja, amely a celofán csomagolásra emlékeztet (ábra. 5). A csecsemők ectropion, eclabium, és az orr és az ízületi porc hypoplasia. A szívást és a pulmonalis lélegeztetést gátolhatják110, a transzepidermális vízveszteséget és a fertőzések kockázatát növelik.110,111

Collodion baba, amely később lamelláris ichthyosis fenotípussá fejlődött.
a Collodion baby A HI and CIE szokásos bemutatása. Autoszomális domináns LI, 112,113 SJ Xhamstergren-Larsson-szindróma,110 trichothyodystrophia,114 juvenilis Gaucher-kór, 110 semleges lipid tárolási betegség, Conradi-H Xhamnermann-Happle-szindróma, Hays-Wells-szindróma és ektodermális dysplasia115 esetenként kollódiumbaba formájában is megjelenhet. A membrán spontán eltűnik az újszülöttek 10-24% – ában, hogy utat engedjen a teljesen normális bőrnek.110 116 a múltban ezeket az eseteket az újszülött LI-ként írták le,117, de nem nevezik SHCB-nek.118 egyes szerzők azért javasolták az önjavító kollodion ichthyosis kifejezést, mert sok ilyen betegnél, amikor később gyermekkorban vagy felnőttként újra megvizsgálják, változó mértékű anhidrosis és hő intolerancia, valamint enyhe ichthyosis jelei vannak, mint például a xerosis és a finom hámlás, különösen a hónaljban és a nyakban.78
sem az optikai mikroszkópia, sem a kollódiumbaba ultrastrukturális vizsgálata nem specifikus. Ezért előnyösebb a bőrbiopsziát a végleges fenotípus kialakulásáig késleltetni.
Shcb-ben szenvedő betegeknél a TGM1,7,119aloxe3,78 és ALOX12B23,78,79 gének mutációit azonosították. Az ALOX12B mutációk a leggyakoribbak. Az SHCB-ben szenvedő 15 skandináv beteg sorozatában 67% – UK mutációt mutatott az ALOX12B génben, 25% – uk az ALOXE3 génben, 8,3% – uk pedig a TGM1 génben.78 mutációt nem találtak néhány betegnél, így más gének is valószínűleg érintettek. Spekulációk szerint ezek a mutációk csökkentik az enzimatikus aktivitást a méhben, de nem a születés után.7 a méhben, ahol a hidrosztatikus nyomás magas, a vízzel történő kelátképzés a mutált enzimet inaktív konformációvá alakítja. Születés után, amikor a nyomás csökken, az enzim visszatér aktív formájába, aktivitása pedig kellően megnő ahhoz, hogy fenntartsa a normális vagy minimálisan érintett fenotípust.7
Acral öngyógyító Collodion Baby
bár a collodion baby az egész testet érinti, az acral régiókra korlátozódó esetekről számoltak be. 1952-ben Finlay et al.A 120 collodion membrán esetet jelentett, amely csak a kezeket és a lábakat érintette, és öngyógyító folyamatot követett. A közelmúltban az acral SHCB új esetéről számoltak be a tgm1 gén mutációival összefüggésben.8 nem ismert, hogy ezek a léziók miért korlátozódnak az acral régiókra, bár az enzimaktivitás helyfüggő szabályozásával kapcsolatos tényezők működhetnek.8
fürdőruha Ichthyosis
fürdőruha ichthyosis először független ARCI változatként jelentették 2005-ben, bár korábban különös eloszlású ichthyosis esetekről számoltak be.121-123 főként Dél-afrikai származású betegeknél mutatták ki,9 bár Európából és mediterrán országokból származó egyéneknél is beszámoltak róla.124 születéskor a betegek generalizált kollodion membránnal rendelkeznek, amely ezután elhagyja a méretezés jellegzetes eloszlását. A törzs, a karok proximális régiója, beleértve a hónaljakat, a nyakat és a fejbőrt, általában érintett, míg az arc középső része, a végtagok és a mellékvese régió általában megkímélte.9 a mérleg nagy, lamellás, sötét színű. Finomabb hámlás fordulhat elő a poplitealis és antecubitalis fossae-ban.124 125 a tenyerek és a talpak enyhe diffúz hyperkeratosisban szenvednek, míg a kezek és a lábak hátsó része nem mutat érintettséget.
az érintett bőrön végzett kórszövettani vizsgálat jelentős hyperkeratosist mutat parakeratosis nélkül, normál szemcsés rétegeket, enyhe vagy közepesen súlyos acanthosist és enyhe lymphocytás infiltrátumot mutat a dermis felső részén.9 az elektronmikroszkópos megfigyelések a legtöbb esetben összhangban vannak a 2. típusú veleszületett ichthyosissal. A nem érintett bőr nem mutat rendellenes eredményeket.124 125 egészséges bőrben a TGase 1 aktivitása enyhén csökken, és általában a pericelluláris területeken lokalizálódik. Az érintett bőrben az enzimaktivitás reziduális és abnormálisan a citoplazmában helyezkedik el.124
mutációkat mutattak ki a tgm1 génben minden eddig vizsgált fürdőruhás ichthyosisban szenvedő betegnél.119,124–126 a leggyakoribb mutáció a P.Arg315Leu, amelyet a legtöbb Dél-afrikai betegnél azonosítottak, és alapító mutáció lehet. Oji et al.124 azt javasolta, hogy a bőr hőmérséklete szerepet játszhat ezeknek a megnyilvánulásoknak a kialakulásában. A digitális termográfia segítségével a szerzők erős összefüggést mutattak a testhőmérséklet és a hámlás között, a test legforróbb területei voltak a leginkább érintettek. Aufenvenne et al.127 A tgase 1 aktivitás optimális hőmérsékletének csökkenését mutatta fürdőruhás ichthyosisban szenvedő betegeknél. Ezt a csökkenést nem figyelték meg egészséges kontrollokban vagy generalizált LI-ben szenvedő betegeknél. Az optimális hőmérséklet a normál enzim esetében 37 C, a mutált enzim esetében 31 C.
kezelés
az ichthyosis kezelésének elsődleges célja a hámlás megszüntetése és a xerosis csökkentése anélkül, hogy túlzott irritációt okozna (3.táblázat). A kezelés eldöntése előtt figyelembe kell venni a beteg életkorát és nemét, a betegség típusát és súlyosságát, valamint a léziók mértékét és helyét.128
terápiás stratégia autoszomális recesszív veleszületett Ichtiózisokban.
| az autoszomális recesszív veleszületett ichthyosis terápiás stratégiája | |
| nátrium-hidrogén-karbonáttal vagy búzakeményítővel, kukoricakeményítővel vagy rizskeményítővel való fürdés és a mérleg mechanikai eltávolítása; a mérleg mechanikus eltávolítása (napi 1 vagy 2 alkalommal) | |
| helyi kezelés (szekvenciális) | Karbamidtartalmú hidratálókkeratinolitikumok propilén-glikolkombinált keratinolitikumokkal (propilénglikol, ons-hidroxi-savak vagy karbamid)keratinolitikumok szalicilsavval kombinálvatopikális retinoidokaz újszülötteknél és a kisgyermekeknél a hatóanyagot nem tartalmazó vivőanyagot kell alkalmazni. Kerülje a karbamidot, a szalicilsavat és a tejsavat a szisztémás felszívódás kockázata miatt |
| orális kezelés | orális retinoidok (acitretin vagy izotretinoin) |
| egyéb intézkedések | az ectropion szemész általi nyomon követésea külső fül rendszeres tisztítása a fül-torok-orr szakember általfizioterápia a kontraktúrák megelőzésére.A megerőltető tevékenységek elkerülése magas környezeti hőmérsékletenhidroterápia |
ARCI-ben szenvedő betegek számára a napi fürdőzés javasolt a mérlegek és a hidratáló nyomok mechanikus eltávolítása érdekében. Ez könnyebb, ha a beteget 15-30-ra vízbe merítikperc. Egyes szerzők azt javasolják, hogy nátrium-hidrogén-karbonátot adjanak a fürdőhöz, hogy denaturalizálják a keratinokat, és lúgossá tegyék a vizet, és így megkönnyítsék a mérleg eltávolítását.129 egyéb hozzáadható termékek közé tartozik a búzakeményítő, a kukoricakeményítő vagy a rizskeményítő. A fürdőolajok nem megfelelőek, mivel elzáródáshoz vezethetnek, ami később a baktériumok elszaporodásának és a termoreguláció romlásának kockázatával járhat.
helyi kezelés
a hidratálók és a helyi keratolitikus szerek általában az első terápiás lehetőség. Javítják a bőr barrier funkcióját és megkönnyítik a hámlasztást. Enyhe helyi káros hatások, például átmeneti viszketés, irritáció vagy szúró érzés fordulhatnak elő.
nátrium-klorid, karbamid, E-vitamin-acetát, glicerin és vazelin használható nedvesítőszerként és kenőanyagként. Vastag hámlasztásban és kifejezett hyperkeratosisban szenvedő betegeknél 1 vagy több keratolitikus szer,mint például a 630 szalicilsav, N-acetilcisztein,131-133 karbamid (>5%), 134 és propilénglikol adható hozzá. A keratinocita differenciálódás modulátorait is használják. Ezek közé tartoznak a lokális retinoidok (tretinoin, adapalén, tazarotén),135,136 kalcipotriol,137 és dexpanthenol.A helyi retinoidok gyakran irritációt és apró, nagyon fájdalmas repedéseket okoznak.137 ezenkívül fennáll a felszívódás és a teratogenitás kockázata a termékeny nőknél, ha túl széles körben alkalmazzák őket.138 a keratolitikumok és hidratálószerek hatékonyságának növelése érdekében az okklúziós kötszer alkalmazható a kezelésnek ellenálló területeken.139 additív vagy szinergikus hatás érhető el 2 vagy több keratolitikus szer vagy hidratálószer kombinálásával is.140-142 a kezelést minden egyes személy számára optimalizálni kell, tekintettel az állapot nagyon változó jellegére, a bőr érzékenységére, valamint az egyes kezelésekre adott válaszreakciók különbségeire. Az optimalizálási folyamatot úgy lehet segíteni, hogy a test egyik oldalát a másiktól eltérően kezeljük az összehasonlítások lehetővé tétele érdekében. Az újszülötteket és a kisgyermekeket hatóanyag nélküli vivőanyaggal kell kezelni, mivel a bőr nagyon finom és érzékeny, és a legtöbb keratolitikum nem tolerálható. Ezenkívül nagyobb a helyi termékek, például a karbamid, a szalicilsav és a tejsav perkután felszívódásának kockázata.143-145
szisztémás kezelés
az orális retinoidok keratolitikus hatásúak, amelyek segítenek eltávolítani a pikkelyeket és megelőzni a túlzott hyperkeratosist. Mind az izotretinoin, mind az aromás retinoidok (acitretin és etretinát) hatásosnak bizonyultak az ARCIs kezelésében.128 146 147 Acitretin 0,5-1 mg/kg/nap dózisban a legszélesebb körben alkalmazott gyógyszer, különösen LI-ben szenvedő betegeknél.148 a CIE-ben szenvedő betegek teljesebb választ adhatnak alacsonyabb dózisok esetén.
a fő mellékhatások a mucocutan betegségek, a teratogenitás, a vázizomrendszeri betegségek és a kóros lipidprofil és a transzaminázszint emelkedés.149-152 a teratogenitás tekintetében az etretinát és az acitretin esetében a terhesség alatt kerülni kell a gyógyszereket, és a betegeknek a kezelés abbahagyása után 3 évig kerülniük kell a terhességet.151 az izotretinoin felezési ideje rövidebb, és 1 hónap elteltével teljesen kiürül a szervezetből, ezért lehet az előnyben részesített lehetőség azoknál a nőknél, akik teherbe kívánnak esni.128
a kezelés ellenőrzése magában foglalja a májfunkciós vizsgálattal és a lipidprofil laboratóriumi vizsgálatát a kezelés megkezdése előtt, majd a kezelés megkezdése után 1 havonta és 3 havonta. Termékeny nőknél terhességi tesztet kell végezni a kezelés megkezdése előtti 2 hétben, és hatékony fogamzásgátló módszert kell alkalmazni a kezelés előtti 4 héttől az azt követő 3 évig (az acitretin esetében). Ha hosszabb ideig tartó retinoid-kezelésre van szükség, ellenőrizni kell a növekedést és a csontfejlődést. Egyes szerzők azt javasolják, hogy a kezelés előtt csontvizsgálatot végezzenek, amelyet éves vizsgálat követ.151 A legújabb irányelvek nem javasolják a rutin radiográfia elvégzését a lehetséges káros hatások miatt.152 ehelyett szelektív radiográfiai vizsgálatokat javasolnak olyan betegeknél, akiknek atipikus csontfájdalmuk van.152
a szisztémás retinoid kezelés alternatívája a retinsav metabolizmusát blokkoló szerekként ismert gyógyszerek alkalmazása, amelyek növelik a retinsav endogén szintjét. Az egyik ilyen gyógyszer a liarozole, amely az Európai Gyógyszerügynökség és az Egyesült Államok Élelmiszer-és Gyógyszerügyi Hivatala li, CIE és HI kezelésére árva státuszt kapott.153-155 ez a gyógyszer a klinikai vizsgálatokban hatékonyabbnak bizonyult, mint az acitretin, és jobban tolerálható és jobb farmakokinetikai profillal rendelkezik.154
egyéb orvosi ellátás
ectropionos betegeknél a mesterséges könnyek és a szem kenőanyagok alkalmazása, valamint az arc és az arc bőrének hidratálása csökkentheti a palpebrális visszahúzódást. A műtéti korrekció súlyos esetekben érvényes lehetőség, de ezt általában néhány évvel később meg kell ismételni. A hidroterápia hasznos lehet.156 beteg figyelmét fel kell hívni arra, hogy kerülje a megerőltető fizikai aktivitást, ha a környezeti hőmérséklet magas, mivel a hypohidrosis hőguta és görcsök kockázatát hordozza magában. Az orális retinoidok javíthatják a hőszabályozást.157 a fizioterápia fontos a hajlítási kontraktúra megelőzésében, különösen a HI esetében. A külső hallójárat rendszeres tisztítása a fül-torok-orr szakember által megakadályozhatja a mérlegek felhalmozódását, így megakadályozhatja a halláskárosodást.
genetikai tanácsadás és prenatális diagnózis
amikor a beteg ichthyosissal diagnosztizálódik, megfelelő genetikai tanácsadást kell felajánlani neki, amelyben elmagyarázzák a rendellenesség természetét, az átviteli módot és a családon belüli jövőbeli megnyilvánulások kockázatát. A prenatális diagnózis jelezheti, hogy a magzat érintett-e, és ha ez a helyzet, a család pszichológiai felkészítése és a terhesség és a szülés során várható problémák merülhetnek fel. A szülőknek lehetőségük van abortuszra, ha nem áll rendelkezésre kezelés. Ezen túlmenően, ha a jövőben elérhetővé válik ezeknek a betegségeknek a génterápiája, a prenatális diagnózis lehetővé tenné ennek a terápiának a lehető legkorábbi alkalmazását.
több mint 20 éve a prenatális diagnózist a magzati bőr biopsziás mintájának vételével és optikai mikroszkópiával, elektronmikroszkópiával vagy immunhisztokémiával tanulmányozták.158 159 ezt az invazív eljárást csak a terhesség késői szakaszában, a terhesség 15.és 23. hete között lehetett elvégezni, és a magzat elvesztésének 1-3% – os kockázatával járt.160 161 az örökletes bőrbetegségek molekuláris mechanizmusainak azonosítása lehetővé tette a genetikai technikákon alapuló sokkal korábbi diagnózist.A 102 162-164 magzati DNS-t a 15.és 20. hét között végzett amniocentézissel vagy a 10. és 12. hét között végzett korionos villus mintavétellel nyerik. A magzati veszteség kockázata ezekkel a technikákkal kevesebb, mint 0,5-1%.165 a fejlesztés egyéb nem invazív módszerei a magzati sejt DNS és a szabad magzati DNS elemzése az anyai keringésben166, valamint a 3 dimenziós ultrahang alkalmazása.167 168
a preimplantációs genetikai diagnózis in vitro megtermékenyítési technikákban is lehetséges, így csak a mutációtól mentes megtermékenyített petesejteket ültetnek be a méhbe, ezáltal a legtöbb esetben elkerülhető az abortusz szükségessége.169
jövőbeli stratégiák az Ichthyosis genetikai kezelésére
bár jelentős előrelépés történt az ichthyosis genetikai diagnosztizálásában, új stratégiákat is folytatnak ezekre a betegségekre.170 a bőr a leginkább hozzáférhető szerv a géntranszfer terápiák számára, ezért az ilyen technikák minimálisan invazívak.171 ugyanakkor a bőrnek olyan egyedi immunológiai tulajdonságai is vannak, amelyek nem kedveznek a transzgenikus termék hosszú távú expressziójának.172 az LI-ben az ex vivo géntranszfer folyamata helyreállította a normál TGM1 expressziót és korrigálta az immunszuppresszált egerek hátára átültetett bőr fenotípusát.173 174 nemrégiben az ABCA12 gén mutációi miatt HI-ben szenvedő betegek tenyésztett keratinocitáinak fenotípusát is helyreállították.3
összeférhetetlenség
a szerzők kijelentik, hogy nincs összeférhetetlenségük.