Frontiers in Pediatrics
- Bevezetés
- hosszú QT-szindróma
- LQTS1
- LQTS2
- LQTS3
- LQTS4
- LQTS5
- LQTS6
- LQTS7
- LQTS8
- LQTS9 és LQTS10
- LQTS11
- LQTS12
- LQTS13
- szerzett LQTS
- elektrokardiográfiás jellemzők a hosszú QT-szindróma három gyakori formájában
- genotípus-fenotípus
- az LQTS klinikai kezelése
- LQTS: Szaúdi perspektíva
- összeférhetetlenségi nyilatkozat
- Köszönetnyilvánítás
Bevezetés
a családi vagy örökletes szívritmuszavarok az aritmiák jelentős százalékát, valamint a hirtelen szívhalál (SCD) okozati összefüggéseit tartalmazzák (1, 2). Az elmúlt két évtizedben a tudósok és a klinikusok hatalmas erőfeszítéseket tettek a veleszületett családi aritmiák bonyolult és összetett mechanizmusainak feltárására (1-8). Az aritmogenezis mechanizmusának megértéséhez ismernünk kell a szív sejtszerkezetének alapjait és elektrofiziológiai tulajdonságait. A szív myocyták a szív fő működő sejtjei, és nagymértékben kapcsolódnak egymáshoz, így az impulzusok gyorsan és egyenletesen terjednek. A cardiomyocytákat egy speciális határ választja el egymástól, amelyet interkalált korongnak neveznek; a gap junction fehérjék, a szív desmoszómák és az ioncsatornák az interkalált korongban helyezkednek el (9). A Gap csomópontok szorosan csomagolt connexinekből állnak, amelyek lehetővé teszik a kis molekulák intercelluláris cseréjét, és lehetővé teszik a gerjesztő áramok áramlását az egyik sejtből a szomszédos sejtbe. A desmoszómák az adherens csomópontokkal együtt felelősek az egyes cardiomyocyták mechanikai kötődéséért. Mindezek a komponensek az interkalált lemezekben egymás után elkülönülnek, és mindegyik komponens egyedülálló funkciót tölt be; az egyik komponens megzavarása befolyásolja a többi komponens működését, ami hajlamosítja a szívet aritmiák kialakulására (9-11). A szív ioncsatornái pórusképző fehérjekomplexek, amelyek feszültség-kapuzott és bonyolultan koordinált Ionos áramok befelé és kifelé történő mozgását biztosítják a sejtmembránokon keresztül, ami elengedhetetlen a szívritmus generálásához és terjedéséhez. A hosszú QT-szindróma (LQTS), a rövid QT-szindróma (SQTS), a Sick sinus-szindróma (SSS), a szívvezetési hiba (CCD), a BrS, a katekolaminerg polimorf kamrai tachycardia (CPVT), a korai repolarizációs szindróma (ERS) és a familiáris pitvarfibrilláció (Af) a jelenleg ismert szívcsatorna-kórok, amelyek a szívritmus generálásához és terjedéséhez kapcsolódó gének egyetlen vagy többszörös hibája miatt fordulhatnak elő.
ebben az áttekintésben kizárólag az LQTS-hez kapcsolódó aritmiákról, annak patofiziológiájáról és a jelenleg rendelkezésre álló klinikai kezelésről írunk le. Úttörő szerepet töltöttünk be a családi szívritmuszavarok genetikai patológiájának tisztázásában Szaúd-Arábiában (1, 12-14), még mindig kardiogenetikai vizsgálatokat folytatunk Szaúd-Arábiában, a felülvizsgálat végén megvitatjuk a jelenleg rendelkezésre álló ismereteket a genetikai és klinikai eredmények több Szaúd-Arábiai családból származnak, akiknek kórtörténetében syncope és SCD volt. Hozzáadjuk a rijádi King Faisal Specialist Hospital and Research Centre (15) csapatának nemrégiben közzétett LQTS-adatait is.
hosszú QT-szindróma
a veleszületett LQTS egy örökletes rendellenesség, amelyet a QT-intervallum meghosszabbítása határoz meg az elektrokardiogramon (EKG). Az LQTS minden formájával rendelkező betegek hajlamosak a kamrai tachyarrhythmiára, torsades de pointes (TdP), amely visszatérő ájuláshoz vagy SCD-hez vezet. Sok esetben az ájulás vagy a hirtelen halál lehet az első és egyetlen megnyilvánulás. Az LQTS világszerte becslések szerint 1 2000 embert érint (16). Az LQTS jellemzője a QT-intervallum meghosszabbítása az EKG-n (a pulzusszámra korrigálva, azaz QTc). A QTc normál értéke férfiaknál 440 ms, nőknél 450 ms. A gyermekek esetében az életkor és a nem függő értékek relevánsak. Az LQTS diagnózisára vonatkozó legújabb konszenzusos ajánlás A következő (17, 18):
1. LQTS-t diagnosztizálnak:
a. LQTS kockázati pontszám jelenlétében 6,5, a QT-megnyúlás másodlagos okának hiányában és / vagy
b. egyértelműen patogén mutáció jelenlétében az egyik LQTS génben vagy
c. A Bazett képletével (QTc) korrigált, szívfrekvenciára korrigált QT-intervallum jelenlétében 500 ms ismételt 12 vezetéses elektrokardiogramon, és a QT-megnyúlás másodlagos okának hiányában.
2. Az LQTS 480 és 499 ms közötti QTc jelenlétében diagnosztizálható ismételt 12 ólom EKG-ban megmagyarázhatatlan syncope-ban szenvedő betegben, a QT-megnyúlás másodlagos okának hiányában és patogén mutáció hiányában.
azoknál a betegeknél, akiknél a QTc időtartama 500 ms volt, szignifikánsan nagyobb kumulatív valószínűsége volt az első ájulásnak, mint azoknál a betegeknél, akiknél a QTc időtartama < 500 ms volt a születéstől a 20 éves korig (19). Azonban azoknál a betegeknél, akiknél 2, 3 és 4 syncope epizódot tapasztaltak, a későbbi syncope epizód kockázata gyakorlatilag azonos volt a szűk vagy megnyúlt QTc időtartamú betegek között (19). Az LQTS molekuláris alapja heterogén, és a mai napig 13 különböző gén mutációját írták le ok-okozati kapcsolatban az LQTS-szel (1-8, 19, 20). Genetikai hibát általában az LQTS-betegek 70% – ában találnak e 13 gén egyikében (1-8, 19, 20). A jelenleg ismert 13 különböző típusú LQTS közül a leggyakoribbak az LQTS1, az LQTS2 és az LQTS3, a szív ioncsatorna génjeinek, a kcnq1, a KCNH2 és az SCN5A hibái miatt. Az összes LQTS okozati mutáció kilencven százaléka ebben a három génben található (1-8, 19, 20). A fennmaradó 10 gén mutációi ritkák, és csak a jelenleg ismert LQTS mutációk 10% – át teszik ki.
a hosszú QT-szindróma általában autoszomális domináns betegség, de alkalmanként egyetlen génben vagy különböző génekben több mutáció is megtalálható az LQTS-ben szenvedő betegek 5-10% – ában (21, 22). A több mutációval rendelkező betegek QTc-je hosszabb lehet, mint az egyetlen mutációval rendelkezőké, és az ilyen betegeknél az életveszélyes kardiális események kockázata is 3,5-szeresére emelkedett (21, 22)
LQTS1
a KCNQ1 gén mutációi az összes LQTS leggyakoribb formája, amelyet LQTS 1-es típusnak (LQTS1) neveznek. Az összes LQTS okozati mutáció ötven százaléka megtalálható a KCNQ1 génben (20). A KvLQT1 (más néven Kv7.1) A KCNQ1 gén által termelt fehérje, és a fehérje tetramereket képez a sejtek belsejében lévő endoplazmatikus retikulumban, amely feltételezhetően összeáll a (KCNE1 által kódolt) mink fehérjével, majd a szív myocyták plazmamembránjába szállítják, ahol lassan aktiváló áramot közvetítenek, amely felgyorsítja az akciós potenciál repolarizációját a szívszövetekben, ez az áram IKs néven ismert (1.ábra). Az LQTS1 ok-okozati KCNQ1 mutációk többnyire missense mutációk, ritka esetekben frameshift mutációk lehetnek a C-terminális régióban (23). Szívritmuszavar a kcnq1 mutációs hordozókban adrenerg hajtások váltják ki, például érzelmi stressz, fizikai erőfeszítés, búvárkodás, úszás (24-26). Azoknál a betegeknél, akiknél a KvLQT1 transzmembrán doménjein mutációk vannak, nagyobb az LQTS-szel kapcsolatos kardiális események kockázata, és nagyobb az érzékenységük a szimpatikus stimulációra (26, 27). A missense mutációkban szenvedő betegek nagyobb kockázatnak vannak kitéve, mint a nem érzékszervi vagy csonkoló mutációkkal rendelkező betegek (27, 28). A 3′-UTR variációja a KCNQ1 génben szintén jelentősen befolyásolja az aritmia érzékenységét, feltehetően azáltal, hogy befolyásolja a gén expresszióját (29). A kcnq1 mutációk miatt ritmuszavarban szenvedő betegek elég jól reagálnak a blokklánc-blokkolókra, de néhány beteg még mindig kevésbé reagálhat, vagy akár rezisztens is lehet erre a gyógyszerre. Egy friss cikkben, Barsheshet et al. (30) azt állította, hogy a citoplazmatikus hurok (c-hurok) régión kívüli mutációkkal rendelkező betegek a KvLQT1-ben kevésbé reagálnak a blokklánc-blokkolókra.
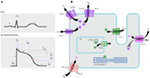
1. ábra. A kamrai akciós potenciálhoz hozzájáruló Ionos áramok (A) és egy olyan kardiomiocita sematikus ábrázolása, amely (csak) az öröklött aritmia szindrómák patogenezisében részt vevő fehérjéket jeleníti meg (B). Az (A) pontban az akciós potenciál igazodik az EKG alatti hozzávetőleges cselekvési idejéhez. A (B) pontban az ankyrin-B-t, a 4.típusú hosszú QT-szindrómában részt vevő adapterfehérjét nem ábrázolják.
a KCNQ1 gén homozigóta vagy összetett heterozigóta mutációi, amelyek a betegség recesszív formáját, az 1-es típusú Jervell és Lange-Nielsen szindrómát (JLNS) okozhatják (31), meglehetősen ritkák. A JLNS-betegek súlyos szívritmuszavarokban és süketségben is szenvednek (31, 32). A JLNS-t okozó KCNQ1 mutációkban szenvedő betegeknél általában nincs funkcionális IKs (28, 31-33). Lehetséges, hogy egyes betegeknél nem lehet süketség annak ellenére, hogy homozigóta vagy összetett heterozigóta mutációk vannak a KCNQ1-ben, az ilyen eseteket autoszomális recesszív LQTS1-nek nevezik (13, 32). Ezeknél a betegeknél kis mennyiségű funkcionális IKs áram (<az összes IKs 10% – a) továbbra is jelen lehet, ami fenntartja a hallásfunkciót, de szívritmus-rendellenességeik ugyanolyan súlyosak, mint a JLNS betegeknél (13, 32).
LQTS2
ez a fajta LQTS ugyanolyan elterjedt, mint az LQTS1, amely a kimutatható mutációval rendelkező LQTS betegek 35-40% – át teszi ki (1, 20, 24, 25). A KCNH2 a HERG fehérjét (Kv11.1) kódolja, amely a gyorsan aktiváló késleltetett egyenirányító k+ áramának (IKr) 6-alegysége. Ebben a génben a Kv11.1 csatornafunkciót csökkentő patogén mutációk meghosszabbítják a QT-intervallum időtartamát (1.ábra), és okozati összefüggést mutatnak az LQTS2-vel. Az LQTS2 szinkopális rohamainak huszonkilenc százaléka pihenés/alvás közben fordul elő, és a szinkopális rohamoknak csak 13% – át jelentették edzés közben (25, 34). Hirtelen megdöbbentő zajok, például ébresztőóra gyűrűk, ajtócsengők, telefoncsengések, jellemzően szinkopális rohamokat váltanak ki ezeknél a betegeknél (24, 34). A Kv11.1 pórusképző régiójában mutációkkal rendelkező betegek (kcnh2 gén által kódolva) hajlamosak az aritmiával kapcsolatos szíves események magas kockázatára, összehasonlítva a nem pórusos régió mutációival rendelkező betegekkel (35). Újszülötteknél a 2: 1 atrioventrikuláris (AV) blokk előnyösen kapcsolódik a KCNH2 mutációkhoz (36). Az LQTS által komplikált teljes AV blokkot a kcnh2 gén mutációjával rendelkező felnőtt betegek 17% – ában is találták (37). A kcnh2 homozigóta mutációi ritkák, és ha jelen vannak, a betegek súlyos LQTS-ben szenvednek, val vel 2:1 AV blokk és súlyos kamrai aritmiák, intrauterin stádiumokban, valamint születés után (11, 38-40). Az LQTS2 patológiában leírt számos mutáció mellett a kcnh2 gén közös polimorf variánsai is modulálhatják a betegség súlyosságát. Érdekes példa a k897t polimorfizmus (SNP) a KCNH2-ben, amely az általános népesség 33% – ában van jelen (41). A jelentések szerint a K897T súlyosbította a kcnh2 mutációk patogenitását (42, 43).
LQTS3
Nav1.Az 5. ábra a feszültségfüggő szív Na+ csatornájának pórusképző fő alegysége, az SCN5A gén által kódolt integrált membránfehérje, amely részt vesz a szív akciós potenciáljának iniciálásában és vezetésében. A szív Na + csatornái egy pórusképző (scn5a által kódolt) és egy vagy több kiegészítő (kiegészítő) alegységből állnak.
az LQTS3-at olyan funkciómutációk nyeresége okozza, amelyek megzavarják a gyors inaktivációt a főcsoporton (1.ábra), és az SCN5A gén mutációját az összes LQTS-ben szenvedő beteg < 10% – ánál írták le mutációval (1, 20, 24). Az LQTS3 betegek szívbetegségeinek többségét (39%) alvás/pihenés közben tapasztalják (25), és az események körülbelül 13% – át jelentették edzés közben (25). Számos esetben egyetlen SCN5A mutációról kimutatták, hogy két vagy akár három különböző aritmia fenotípust fejt ki ugyanabban a családban, például LQTS, BrS vagy CCD (44-47). Az LQTS3 mutációval rendelkező férfi betegek tünetei sokkal korábban alakulhatnak ki, mint a női betegek (48). Mind a heterozigóta, mind a homozigóta SCN5A mutációkat az LQTS3 leírta funkcionális 2:1 AV blokkkal (49). Nemrégiben találtunk egy iráni családot 1507_1509delqkp mutációval több családtagban, ahol a betegek LQTS-t és CCD-t kombináltak (az adatok nem jelennek meg). Ezt a mutációt más országokból származó betegeknél is jelentették, ami arra utal, hogy ez egy visszatérő és hot spot mutáció . 1493delk és 1795insd mutációk (44, 51) is beszámoltak az akciós potenciál különböző fázisaiban ilyen funkcióvesztési és funkciónövekedési jellemzőkkel rendelkező mutációkról.
esetenként az SNP patogén hatást is gyakorolhat hordozóira. Az S1103Y az SCN5A gén gyakori változata, amely az afroamerikaiak 13% – ában van jelen (52). Az ezzel a variánssal rendelkező hordozók fokozottan ki vannak téve az aritmiák és a hirtelen csecsemőhalál szindróma (SIDS) kockázatának (52).
LQTS4
az LQTS4 az LQTS első nem csatornás formáját jelenti. Az ank2, egy adapterfehérje mutációja intracelluláris kalcium túlterheléshez vezet, amely hozzájárul az LQTS4-hez (53, 54). A QT-megnyúlás mellett az ebben a szindrómában szenvedő betegek sinus bradycardia, paroxysmal AF és CPVT (54) lehetnek. Az ANK2 mutációk patogén hatása közepes vagy súlyos lehet, és a klinikai tünetek a mutáció súlyosságától függenek.
LQTS5
a Kcne1 mutációi az LQTS5-hez kapcsolódnak (1.ábra) (55, 56). A heterozigóta KCNE1 mutációk csökkentik az IKs-t azáltal, hogy domináns negatív hatást gyakorolnak a kísérő normál allélra, és késleltetett szív repolarizációhoz vezetnek (1.ábra), amely felelős az aritmiák fokozott kockázatáért (6). A homozigóta kcne1 mutációkkal rendelkező betegek JLNS-ben szenvednek (2.típus) (57, 58).
a D85N polimorfizmus a KCNE1 génben, jelen van a 0-ban.Az általános népesség 7-1% – a (41). Egy tanulmány szerint Nishio et al. (59), A d85n polimorfizmust gyakrabban találták meg az LQTS-betegeknél, ami kockázati genotípust jelent az LQTS-patológiában (potenciálisan csak az ázsiai populációban). Európában a d85n-t a szerzett LQTS (aLQTS) betegek 5% – ánál jelentették (32 betegből álló kohorszban), akik TDP-t tapasztaltak (60).
LQTS6
a KCNE2 gén a mink-rokon peptid 1-et (MiRP1) kódolja, amely a kardiális káliumcsatorna IKR feltételezett ons alegysége (1.ábra). A kcne2 gén mutációi a késleltetett egyenirányító káliumáram (IKr) gyorsan aktiváló komponensének hibáihoz is vezethetnek, az LQTS6 patológiás alapja (61). Auditív / akusztikus inger, például ébresztőóra zaj, csengő stb. szinkopális támadásokat válthat ki a KCNE2 mutációs hordozókban, hasonlóan a KCNH2 mutációhoz (62).
LQTS7
ez a szindróma Andersen–Tawil szindróma (ATS) néven is ismert. Az ATS ritka rendellenesség, amelyet alkalmi szinkopás, szívmegállás jelent. Az EKG jellemzői közé tartozik az enyhe QT-intervallum megnyúlása, rendellenes U hullámok, gyakori kamrai ektópia, kétirányú kamrai tachycardia (VT), polimorf VT. Ez a szindróma extrakardiális funkciókat is mutat, például vázizom periodikus bénulás és fejlődési problémák, mint például a szájpadlás, az alacsony fülek, a rövid testtartás és a végtagok fejlődési rendellenességei (63). A klinikailag diagnosztizált ATS-betegek többségéről beszámoltak a kcnj2 mutációjáról (63). A KCNJ2 a befelé egyenirányító káliumcsatornák pórusképző alegységét kódolja (IK1) (1.ábra) (64, 65).
LQTS8
más néven Timothy-szindróma (TS), a betegek súlyos QT-megnyúlást mutatnak EKG-jükön, amelyet szindaktikusan, születéskor kopaszsággal és kis fogakkal kombinálnak az esetek 100% – ában, és kevésbé penetráns szív szerkezeti rendellenességek, mentális retardáció, autizmus és arc diszmorf funkciók (66). Két altípusa van: ts1 (klasszikus) és TS2 (ritka forma).
a TS2 kardiológiailag keményebb, mint a TS1 (66, 67). A TS2 betegeknél szintén hiányzik a syndactyly (67). Mutációk a cacna1c gént kódoló L-típusú kalciumáram (ICa-L) 6 alegységében a TS (LQTS8) mindkét formájához vezetnek. A cacna1c génben vannak alternatív módon összekapcsolt, egymást kölcsönösen kizáró exon-8 exon-8 és exon-8A. a szívben és az agyban, ahol a CACNA1C túlnyomórészt expresszálódik, az exon-8 az mRNS transzkriptumok 80%-ában, az exon-8a pedig 20% – ban van jelen. (66). A G406R mutáció a 8a exonban ok-okozati a TS (TS1) klasszikus formájához képest, a G402S-t pedig a 8 exonban a SEVERER-ben a TS2-ben jelentették. A lista új kiegészítése az Ala1473Gly mutáció, amelyet egy tovább kibővített fenotípusú TS csecsemőben írtak le (68). Minden mutáció vagy de novo vagy mosaic volt a szülőben, és az ICA-L csatorna működésének összes eredménye (66-69).
LQTS9 és LQTS10
a CAV3 vagy SCN4B mutációi a késői INa-ban funkciónövekedést eredményeznek, ami LQTS3-szerű fenotípust (70-74) okoz. Ezek LQTS9 (cav3 mutációval társítva) és LQTS10 (scn4b mutációval társítva) néven ismertek.
Caveolae szépen leírt Engelman et al. (72) mint” kis barlangok ” a plazmamembránban. Ezek kis bevonat nélküli gödrök, és fontos dinamikus és szabályozó események helyszínének tekintik a plazmamembránon (72, 73). A caveolinok a caveolae fő fehérjéi, és a caveolin-3 (A CAV3 gén által kódolt) kifejezetten a cardiomyocytákban és a vázizomsejtekben található. Számos szívioncsatornáról számoltak be, amelyek a caveolin-3-ban dúsított szív myocytákból kivont caveolae-ban lokalizálódnak (72, 73). Továbbá, komponensei a caiveolae-dúsított membránokban (72, 73) jelen vannak a caiveolae receptor jelátviteli kaszkád.
az SCN4B kódolja a NAV-ot a 4-hez, amely a szív nátriumcsatornájának egy kisegítő főegység-alegysége. Eddig csak egy mutációt (L179F) jelentettek ebben a génben egy mexikói családban, több érintett családtaggal (74). Megállapították, hogy ebben a génben a mutáció a Nav1.5 áram funkciójának növekedését eredményezi (74).
LQTS11
a szívben a szív akciós potenciáljának időtartamának szimpatikus szabályozását (APD) a következők közvetítik: ~ -adrenerg receptor (~- AR) aktiváció, amely megköveteli az AKAP9 (Yotiao) összeszerelését az IKS csatorna ~ -alegységével (KvLQT1). Az AKAP9 mutációja LQTS11-et okoz (75). A mai napig csak egy mutációról, az S1570L-ről számoltak be az AKAP9-ben (75).
LQTS12
mutáció a 6-1-Syntrophin (SNTN1) gén okozati LQTS12. Ebben a génben a mutáció a szív nátriumcsatorna (Nav1.5) funkciójának növekedéséhez vezet, amely az LQTS12 (76) kóros alapja.
LQTS13
a G fehérjéhez kapcsolt, befelé egyenirányító káliumcsatorna alegységet (Kir3.4) a KCNJ5 gén kódolja. A funkcióvesztés mutációja ebben a génben LQTS13-at okozhat (77). Eddig csak egy mutációt, a G387R-t írtak le egy kínai családban, ahol kilenc beteg rendelkezik ezzel a mutációval. A Kir3.4 csökkent plazmamembrán expresszióját javasolták az LQTS patológiájaként a betegeknél.
szerzett LQTS
a veleszületett LQTS mellett az LQTS alqts néven ismert másik változata is létezik, amelyet olyan tényezők és anyagok okoznak, amelyek csökkentik a kálium fluxust és rontják a szívizom repolarizációs képességét. Jól elismert állapotok a női nem, a hypokalemia és a szív káliumcsatornáit gátló gyógyszerek (78-80). Számos általánosan felírt gyógyszer előnyösen megkötheti és blokkolhatja a HERG csatornát (Kv11.1, A kcnh2 gén által kódolt fehérje), és hajlamos az aLQTS-re (79, 80). Nemrégiben kimutatták, hogy az IKs blokádja hozzájárulhat a gyógyszer által kiváltott aLQTS-hez is, különösen akkor, ha a repolarizációs tartalék veszélybe kerül (81). Megállapították, hogy a fluoxetin és a norfluoxetin elnyomja az IKs tulajdonságait, mind in vivo, mind in vitro, és jelentős LQTS-hez vezetett (81). A jelentések szerint a polimorfizmusok, a d85n a minkben (kcne1 gén), a T8A, a Q9E a MiRP1-ben (kcne2 gén), amelyek az IKs és IKr csatornák feltételezett 6-alegységei, aLQTS-t okoznak (82, 83). Autoimmun LQTS-ről is beszámoltak egy olyan beteg esetében, akinek IgG-je anti-HERG antitesteket tartalmazott (84).
elektrokardiográfiás jellemzők a hosszú QT-szindróma három gyakori formájában
tipikus ST-T-hullám minták vannak jelen a genotipizált LQTS-betegek többségében, és felhasználhatók az LQTS1, LQTS2 és esetleg lqts3 genotípusok azonosítására (85, 86).
az LQTS lqts1 formája széles T-hullámmal társul, anélkül, hogy a QT-intervallum lerövidülne edzés közben (2a ábra). Az LQTS2 alacsony amplitúdójú, gyakran bifid, T hullámokkal társul (2b ábra). Az LQTS3 egy hosszú iso-elektromos szegmenshez és egy keskeny alapú, magas T hullámhoz kapcsolódik (2C ábra). A TDP kezdetének szüneteltetése veleszületett LQTS-ben genotípus-specifikus, az lqts2-ben domináns, de az LQTS1-ben szinte hiányzik (87).

2. ábra. Elektrokardiogram felvételek lqts1, LQTS2 és LQTS3 betegektől. (A) 12-ólom EKG egy 18 éves férfi egy KCNQ1 mutáció. A QT-intervallum megnyúlik (QTc = 500 ms). Az ST szegmens széles bázissal és viszonylag nagy amplitúdóval rendelkezik. A vezetési intervallum normális (standard kalibrálás). (B) 12-ólom EKG egy 14 éves lány egy KCNH2 mutáció. A QT-intervallum megnyúlik(QTc 520 ms). Az ST szegmens a v3 ólomban van bevágva, viszonylag alacsony amplitúdóval rendelkezik a végtagi vezetékekben. A vezetési intervallum normális (standard kalibrálás). (C) 12-ólom EKG egy 12 éves fiú egy SCN5A mutáció. A QT-intervallum megnyúlik(QTc 600 ms). Az ST szegmensnek hosszú (majdnem) iso-elektromos szegmense van, nagy, éles és keskeny T hullámmal. A vezetési intervallum normális (standard kalibrálás).
bár a minták az LQTS specifikus genotípusára utalhatnak, gyakran találtak kivételeket.
genotípus-fenotípus
a hosszú QT-szindróma autoszomális domináns betegség. A heterozigóta mutációhordozók genotípus-és fenotípus-elemzését az LQTS1, LQTS2 és LQTS3 betegeknél meglehetősen széles körben végezték. Gyermekkorban a cardialis események kockázata szignifikánsan magasabb az LQTS1 férfiaknál, mint az lqts1 nőknél, míg az lqts2 és LQTS3 betegeknél (88-91) nem figyeltek meg szignifikáns, nemhez kapcsolódó különbséget a cardialis események kockázatában. Felnőttkorban (40 éves kor után is) az LQTS1 és LQTS2 nőknél szignifikánsan magasabb lehet a kardiális események kockázata, mint a férfiaknál (88-91). Általánosságban úgy tűnik, hogy az LQTS3 betegeknél a kardiális események letalitása dominál, mint az LQTS1 és LQTS2 betegeknél (91). Az LQTS-ben szenvedő nőknél csökkent a szívbetegségek kockázata a terhesség alatt, de a kockázat meglehetősen növekszik a 9 hónapos szülés utáni időszakban, különösen a kcnh2 gén mutációjával rendelkező nőknél (92).
a gyermekek hirtelen szívhalálát a szív ioncsatorna génjeinek mutációi is okozhatják (93-96). A megmagyarázhatatlan SCD-ben szenvedő gyermekek körülbelül 28% – A (1 és 18 év között, átlagéletkor: 12,3 3,8 év) mutációk hordozói voltak az LQTS ok-okozati génjeiben (97). A SIDS-ben az SCN5A mutációi dominánsnak tűntek (98, 99), de a KCNQ1, KCNH2, KCNE2 és CAV3, SCN4B és SCN3B mutációkat is találtak (100, 101). Méhen belüli magzati halálesetekről is beszámoltak a szív ioncsatorna génjeinek hibái miatt (12, 102).
az LQTS klinikai kezelése
minden olyan gyógyszer abbahagyása, amelyről ismert, hogy meghosszabbítja a QT-intervallumot, valamint az elektrolit-egyensúlyhiány korrekciója és/vagy az anyagcsere-állapotok kiváltása legyen az elsődleges hangsúly a (szerzett) LQTS betegek kezelése során. Az LQT-k tünetei gyakran adrenergikusan közvetítettek, ezért általában ajánlott a betegek atlétikai tevékenységekben való részvételének korlátozása (24, 25). Az LQTS klinikai terápiájának alapja a blokád. A propranolol, a nadolol és a metoprolol hosszú hatású készítményeit általában használják, és hatékonyságukat a blokádban a testmozgás pulzusának tompításával értékelik (például >20%) (24, 25). Az összes közül a inhibitorok, propranolol és nadolol tartják jobb, mint a metoprolol tüneti betegek (103). Továbbá, az Alzheimer-blokád profilaktikus kezelésként is alkalmazható néma mutációs hordozókban az SCD csökkentésére (24). Egy tanulmányban Barsheshet et al. (30) azoknál a betegeknél, akiknél a KCNQ1 gén C-loop missense mutációi voltak, nagy volt az életveszélyes kardiális események kockázata, és jelentős előnyeik voltak a kezelés során. Mivel az LQTS2-ben szenvedő nőknél fokozott a kockázat a 9 hónapos szülés utáni időszakban, a magas kockázatú időszak alatt a szívbetegségek csökkentése érdekében a 62. Beültethető cardioverter-defibrillátorok (ICD-k) alkalmazása megfontolandó azoknál a betegeknél, akiknél a kezelés ellenére ismétlődő ájulás áll fenn, vagy akiknél magas a szívmegállás kockázata (pl., tüneti LQTS2 és LQTS3 dokumentált QTc-megnyúlással). A bal szív szimpatikus denervációja (LCSD) ajánlott a magas kockázatú LQTS-betegek számára, akiknél az ICD ellenjavallt vagy elutasított, és a CA-blokkolók vagy nem hatékonyak, nem tolerálják, nem fogadják el vagy ellenjavallt (18, 104). A jlns-ben szenvedő betegek QTc-je általában >500 ms, és szintén magas kockázatnak vannak kitéve, az olyan betegeknél, akiknél korai ICD-kezelést javasoltak, nem megfelelő a hatékonyság (18, 31).
LQTS: Szaúdi perspektíva
a Szaúd-Arábiából származó LQTS-ről szóló első jelentést 1993-ban tették közzé a rijádi fegyveres erők Kórházából (105). Négy csecsemőt és 6-48 hónapos kisgyermekeket, akiknek kórtörténetében visszatérő rohamok voltak, egyetlen családból diagnosztizáltak LQTS-t (105). A családtörténetből kiderült, hogy két másik nagycsaládosnak hasonló epizódjai voltak hirtelen eszméletvesztéssel, három családtag pedig hirtelen meghalt (105). A kezdeti diagnózis minden esetben epilepszia volt (105). Néhány évvel később két szórványos esetjelentést jelentettek az újszülött LQTS viszonylag súlyosabb változatával, 2:1 AV blokkkal kombinálva (106, 107). Mindezek a közzétett klinikai jelentések nem voltak olyan genetikai eredmények, amelyek megmagyarázhatnák az LQTS patofiziológiáját bennük (105-107).
először jelentettünk genetikai hibákat az LQTS kóros alapjaként Szaúd-Arábiából származó hasonló betegeknél. Hat Szaúdi családot vizsgáltunk, akiknek kórtörténetében ájulás és hirtelen megmagyarázhatatlan haláleset történt magzat, újszülöttek és gyermekek (12-14). Autoszomális recesszív LQTS1-et két családból származó gyermekeknél diagnosztizáltak (3.ábra). Az autoszomális recesszív LQTS2-t két családban diagnosztizálták (4.ábra). Egy családban egy női beteget autoszomális domináns LQTS2-vel diagnosztizáltak (5.ábra), a betegnek szinkopális rohamai voltak a szülés utáni gyógyulási időszakban a kórházban, ami nagyon gyakori a KCNH2 mutációhordozó nőknél. Minden betegünkben a patogén mutációk azonosítása az LQTS ok-okozati szívioncsatorna génjeiben megerősített klinikai diagnózishoz vezetett, amelyeket tévesen diagnosztizáltak epilepsziás görcsökként, mielőtt hozzánk utaltak volna (13, 14) nemrégiben, Shinwari et al. (15) A King Faisal Szakkórház és Kutatóközpont lqts1 ok-okozati KCNQ1 mutációt, H258P-t jelentett egy nagy családban, ahol 12 érintett személy volt. Csak két hordozó volt tünetmentes, QTc – jük >500 ms volt, és az egyik betegnél a klinikai tüneteket a CA-blokkolók elnyomták, a másiknál pedig ICD-t kellett alkalmazni.
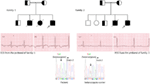
3. ábra. A család törzskönyvi rajza-1 és 2 autoszomális recesszív LQTS1, a probandok nyíllal jelennek meg. A két család probandusaiból származó EKG-k középen láthatók, QTc-jük 557, illetve 529 ms. Intronikus mutáció, c. 387 -5 T > A A kcnq1 génben a betegeknél (alul látható). A mutációt nyíllal mutatták ki. A nem kitöltött körök és négyzetek nem hordozói a mutációnak. Az érintett személyeket kitöltött körök (nők) és négyzetek (férfiak) mutatják. A félig kitöltött négyzetek és körök heterozigóta mutációval rendelkező egyének. Az elhunytakat perjelekkel, a probandusokat nyíllal, a vér szerinti házasságot pedig = Exon − intron határral jelöljük, az exon felé mutató nyíllal ellátott szaggatott vonallal.
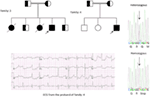
4. ábra. A család törzskönyvi rajza-3 és 4 autoszomális recesszív LQTS2. A proband EKG-je a family-4-ből a törzskönyv rajz alatt látható, amely sinus tachycardiát, szinte teljes AV blokkot, széles komplex menekülési ritmust mutat, nagyon hosszú QTc-intervallumokkal (QT >600 ms). Mutáció, c.3208 C > T (p. Q1070X) a kcnh2 génben a betegeknél (a jobb oldalon látható), nyíllal jelölve.
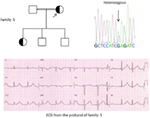
5. ábra. Bal felső sarokban: a család törzskönyve-5. Az érintett egyént kitöltött kör (női) Proband jelzi egy nyíl. 12-a proband vezető EKG-je (I:2). Az EKG magas csúcsú, széles alapú, nagy amplitúdójú T hullámot mutat, a QTc intervallum 580 ms. a KCNH2 gén szűrése a “G” nukleotid helyettesítését mutatja egy “A” – ra (c.2362g > a, nyíl jelölt), ami aminosav-szubsztitúcióhoz vezet, p. E788K.
a Szaúd-Arábiából származó tanulmányunk (12-14) genetikai és klinikai eredményei több okból is érdekesek: (1) összesen hat családot vizsgáltunk, köztük négy homozigóta / összetett heterozigóta volt a mutációk szempontjából, és a mutációk egy ősi forrásból származnak; (2) az LQT-k összes mutációja újszerű volt, csak ezekben az Arab családokban számoltak be; (3) a mutációk homozigozitása vagy összetett heterozigozitása miatt a klinikai fenotípusok szintén súlyosak voltak vizsgált családjainkban (12-14). Azt javasoltuk, hogy a genetikai és fenotípusos megfigyelések A szaúd-arábiai rokonsági házasságok rendkívül magas arányából erednek (108, 109). Tanulmányunk az első tudományos bizonyítékot szolgáltatta a rokonság szerepéről a megmagyarázhatatlan szívritmuszavarok és SCD-k kulcsszerepében Szaúd-Arábiában (12-14). Azt is kimutattuk, hogy a kcnq1 mutáció c.387 -5 T > A (NM_000218) (3 .ábra) Szaúd-Arábia Assir tartományában terjedt el egy közös őstől több generáció alatt a rokonsági házasságok magas előfordulása miatt. Eddig ez a leggyakoribb LQTS1 okozati mutáció a szaúd-arábiai populációban, amelyet a King Faisal Szakkórházban és a Khamis Mashayt katonai kórházban is megfigyeltek (nem publikált). Hasonló eredményt kaptunk az LQTS2 ok-okozati mutáció esetében is, c.3208 C > T (p.Q1070X) (4. ábra) a KCNH2 génben (12, 14), amely Szaúd-Arábiában is alapító mutáció, és az Assir régióban sok generáció alatt elkülönült (lásd a térképet, 6.ábra). Azt jósolnánk, hogy jelentős számú olyan egyén létezik, akiknek az említett ősi/alapító mutációi vannak (mind a KCNQ1, mind a KCNH2 és más gének) ebben a régióban, valamint a nagyvárosokban, mint Rijád, Dzsidda és Dammam a városi migráció miatt. Az LQT-kre patogén több alapító vagy ősi mutációt nagyon terveznek Szaúd-Arábia más tartományaiban a rokon házasságok magas aránya miatt.
6. ábra. Szaúd-Arábia térképe (jóvoltából: Wikipedia). Az Assir régió vastag vonallal van jelölve, ahol a kcnq1 és a KCNH2 gének alapító mutációit találtuk az 1-4 családban.
mivel az LQTS autoszomális domináns betegség, arra számítottunk, hogy túlnyomórészt heterozigóta mutációhordozó betegeket fogunk megfigyelni. De tanulmányunkban elsősorban recesszív mutációkkal rendelkező betegeket azonosítottunk (12-14). Nyilvánvaló, hogy ezek a betegek lesznek az első tünetek, és túlnyomórészt a Prince Sultan Szívközpontba utalták őket, figyelmünk alá vonva őket. Mindazonáltal a vizsgálataink eredményei arra utalnak, hogy a recesszív LQT-k halálos klinikai fenotípusokat mutathatnak a gyermekeknél, és nem ritkák Szaúd-Arábiában (12-14). Mivel a heterozigóta mutáció hordozói szintén hajlamosak az aritmia és szövődményeinek kialakulására, összehangolt kezdeményezést kell tenni, hogy a helyi általános orvosok, kardiológusok és klinikai genetikusok közös platformot hozzanak létre a veszélyeztetett egyének azonosítására. Továbbá jelenleg nincs genetikai információnk más családi aritmia rendellenességekről, például CPVT, SQTS, BrS, AF stb. A speciális kardiogenetikai központoknak kezdeményezniük kell a genetikai hibák, mutációk felkutatását, valamint genotípus-fenotípus vizsgálatokat kell végezniük az örökletes aritmiák minden formájában. Azt is figyelembe kell venni, hogy nem minden genetikai ritmuszavarnak lenne családi anamnézise, mivel sok esetben az aritmia ok-okozati mutációja de novo eredetű, azaz a proband az első beteg a családban a mutációval, és ő a forrás a mutáció továbbítására a downstream generációkban (110, 111). Szaúd-Arábiában a rokonsági házasságok nagyon magas aránya miatt, arra számítunk, hogy sokkal több alapító mutáció döntő szerepet játszik a veleszületett aritmiákban ebben az országban. Ezeknek az alapító mutációknak az azonosítása legyen az első és legfontosabb feladatunk, ami megkönnyítené számunkra a hatékony házasság előtti és tüneti genetikai tanácsadás kidolgozását ebben az országban. Az LQTS okozati génjeinek mutációi a klinikai penetráció változékonyságát eredményezhetik, az egyik végletben egyes hordozók feltehetően teljesen egészségesek lehetnek, de egyes hordozóknak a betegség első megnyilvánulása lehet ájulás vagy hirtelen halál. Egy kisgyermek vagy felnőtt hirtelen halála nagy lelki és érzelmi terhet ró a családra, a hordozók szűrése elengedhetetlen, mivel léteznek egyszerű gyógyszerek, pl. a .. – blokkolók (viselkedésmódosítás is), amelyek nagyon hatékonyan megakadályozhatják a hordozókat az aritmia és az SCDs végzetes következményeitől. Az LQTS ok-okozati génjeiben homozigóta mutációkkal rendelkező egyéneknek súlyos morbiditása és rendkívül magas halálozási aránya lehet, beleértve a magzati brady-tachyarrhythmiákat, és sok esetben vetélést, spontán abortuszt a terhes anyáknál (12-14).
Szaúd-Arábiában nem sok kutatást végeztek a különböző SNP-k előfordulásáról az aritmia kapcsolt génjeiben, valamint az őket szabályozó génekben. Splawski és munkatársai. (112) leírt egy közös s1103y variánst az SCN5A génben, amely az afro-amerikaiak aritmiájához kapcsolódik. Az Y1103 nevű variáns allél felelős a csatorna aktiválásának felgyorsításáért, ezáltal növelve a szívritmuszavarok valószínűségét afrikai származású személyeknél (52, 112). A K393N a KCNQ1 gén egyik változata, amelyet LQTS1 betegeknél jelentettek az USA-ban, de az Arab populációban ezt a változatot az egyének 2% – ában észleltük (nem publikált adatok). Függetlenül attól, hogy a K393n variáns a Kcnq1 génben az Arabokban összehasonlítható-e az afroamerikaiak S1103Y (SCN5A) variánsával vagy a japán lakosság D85N (KCNE1) variánsával, szintén érdemes lehet vizsgálatot végezni (59).
összeférhetetlenségi nyilatkozat
a szerzők kijelentik, hogy a kutatást olyan kereskedelmi vagy pénzügyi kapcsolatok hiányában végezték, amelyek potenciális összeférhetetlenségnek tekinthetők.
Köszönetnyilvánítás
őszinte hálánk Prof. Arthur Wilde-nak, Prof. Connie Bezzinának és a “Heart” folyóirat kiadójának, hogy engedélyezték az 1.ábra használatát ebben a kéziratban Ref. (113).
1. Bhuiyan ZA. Az örökletes szívritmuszavar szindrómák klinikai és genetikai spektruma. Amszterdam: Amszterdami Egyetem (2009).
2. Bossaert L, Breithardt G, Brugada P, et al. Hirtelen szívhalál munkacsoport, Európai Kardiológiai Társaság. Europace (2002) 4:3-18. doi: 10.1053 / eupc.2001.0214
CrossRef Teljes Szöveg
3. Wang Q, Shen J, Splawski I, Atkinson D, Li Z, Robinson JL, et al. Scn5a mutációk, amelyek örökletes szívritmuszavarhoz, hosszú QT szindrómához kapcsolódnak. Cell (1995) 80:805-11. doi:10.1016/0092-8674(95)90359-3
Pubmed absztrakt / Pubmed teljes szöveg / CrossRef teljes szöveg
4. Curran én, Splawski én, Timothy KW, Vincent GM, Zöld ED, Keating MT. A szívritmuszavar molekuláris alapja: a HERG mutációk hosszú QT-szindrómát okoznak. Cell (1995) 80:795-803. doi:10.1016/0092-8674(95)90358-5
Pubmed Absztrakt / Pubmed Teljes Szöveg / CrossRef Teljes Szöveg
5. Wang Q, Curran ME, Splawski I, Burn TC, Millholland JM, Vanraay TJ, et al. Egy új káliumcsatorna gén helyzeti klónozása: a KVLQT1 mutációk szívritmuszavarokat okoznak. Nat Genet (1996) 12:17-23. doi: 10.1038 / ng0196-17
Pubmed absztrakt / Pubmed teljes szöveg / CrossRef teljes szöveg
6. Splawski I, Tristani-Firouzi M, Lehmann MH, Sanguinetti MC, Keating MT. A hmink gén mutációi hosszú QT szindrómát okoznak és elnyomják az IKs működését. Nat Genet (1997) 17:338-40. doi: 10.1038 / ng1197-338
Pubmed absztrakt / Pubmed teljes szöveg / CrossRef teljes szöveg
7. Sanguinetti MC, Jiang C, Curran én, Keating MT. Mechanisztikus kapcsolat az öröklött és a szerzett szívritmuszavar között: a HERG az IKr káliumcsatornát kódolja. Cell (1995) 81:299-307. doi:10.1016/0092-8674(95)90340-2
Pubmed absztrakt / Pubmed teljes szöveg / CrossRef teljes szöveg
8. Neyroud N, Tesson F, Denjoy I, Leibovici M, Donger C, Barhanin J, et al. A kvlqt1 káliumcsatorna gén új mutációja okozza a Jervell és Lange-Nielsen cardioauditory szindrómát. Nat Genet (1997) 15:186-9. doi: 10.1038 / ng0297-186
Pubmed absztrakt / Pubmed teljes szöveg / CrossRef teljes szöveg
9. Kucera JP, Rohr S, Rudy Y. a nátriumcsatornák lokalizálása az interkalált lemezekben modulálja a szívvezetést. Circ Res (2002) 91:1176-82. doi:10.1161 / 01.RES.0000046237. 54156. 0 a
Pubmed absztrakt / Pubmed teljes szöveg / CrossRef teljes szöveg
10. Oxford EM, Musa H, Maass K, Coombs W, Taffet SM, Delmar M. Connexin43 átalakítás, amelyet a plakophilin-2 expresszió gátlása okoz a szívsejtekben. Circ Res (2007) 101:703-11. doi: 10.1161 / CIRCRESAHA.107.154252
Pubmed Absztrakt / Pubmed Teljes Szöveg / CrossRef Teljes Szöveg
11. Rohr S. molekuláris áthallás az interkalált korong mechanikai és elektromos csomópontjai között. Circ Res (2007) 101:637-9. doi: 10.1161 / CIRCRESAHA.107.161901
CrossRef Teljes Szöveg
12. Bhuiyan ZA, Momenah TS, Gong Q, Amin AS, Ghamdi SA, Carvalho JS, et al. Visszatérő méhen belüli magzati veszteség a herg közeli hiánya miatt: homozigóta nonszensz herg Q1070X mutáció klinikai és funkcionális jellemzése. Szívritmus (2008) 5:553-61. doi: 10.1016 / j.2008.01.020
Pubmed Absztrakt / Pubmed Teljes Szöveg / CrossRef Teljes Szöveg
13. Bhuiyan ZA, Momenah TS, Amin TS, Al-Khadra AS, Alders M, Wilde AAM, et al. A 2. exon hiányos kihagyásához vezető Intronikus mutáció a KCNQ1-ben megmenti a hallást a Jervell és a Lange-Nielsen szindrómában. Prog Biophys Mol Biol (2008) 98:319-27. doi: 10.1016 / j. pbiomolbio.2008.10.004
Pubmed Absztrakt / Pubmed Teljes Szöveg / CrossRef Teljes Szöveg
14. Bhuiyan ZA, Al-Shahrani S, Al-Khadra AS, Al-Ghamdi S, Al-Kalaf K, Mannens MMAM, et al. A hosszú QT-szindróma klinikai és genetikai elemzése hat család gyermekeiben Szaúd-Arábiában: különböznek egymástól? Pediatr Cardiol (2009) 30:490-501. doi: 10.1007 / s00246-008-9377-y
Pubmed absztrakt / Pubmed teljes szöveg / CrossRef teljes szöveg
15. Shinwari ZM, Al-Hazzani A, Dzimiri N, Tulbah S, Mallawi Y, Al-Fayyadh M, et al. Új KCNQ1 mutáció azonosítása egy nagy Szaúdi családban, hosszú QT-szindrómával: klinikai következmények és megelőző következmények. Clin Genet (2013) 83:370-4. doi: 10.1111 / j. 1399-0004.2012. 01914.x
Pubmed absztrakt / Pubmed teljes szöveg / CrossRef teljes szöveg
16. Schwartz PJ, Stramba-Badiale M, Crotti L, Pedrazzini M, Besana A, Bosi G, et al. A veleszületett hosszú QT szindróma előfordulása. Keringés (2009) 120: 1761-7. doi: 10.1161/CIRCULATIONAHA.109.863209
Pubmed Absztrakt / Pubmed Teljes Szöveg / CrossRef Teljes Szöveg
17. Schwartz PJ, Moss AJ, Vincent GM, Crampton RS. A hosszú QT-szindróma diagnosztikai kritériumai. Frissítés. Keringés (1993) 88:782-4. doi:10.1161 / 01.CIR.88.2.782
CrossRef Teljes Szöveg
18. A Priori SG, Wilde AA, Horie M, Cho Y, Behr ER, Berul C, et al. Összefoglaló: HRS / EHRA / APHRS konszenzus nyilatkozat az örökletes primer arrhythmia szindrómában szenvedő betegek diagnózisáról és kezeléséről. Europace (2013) 15:1389-406. doi:10.1093 / europace / eut272
CrossRef teljes szöveg
19. Liu JF, Jons C, Moss AJ, McNitt S, Peterson DR, Qi M, et al. Szindróma Nyilvántartás. A visszatérő syncope és az azt követő fatális vagy majdnem fatális események kockázati tényezői hosszú QT-szindrómás gyermekeknél és serdülőknél. J Am Coll Cardiol (2011) 57:941-50. doi: 10.1016 / j.jacc.2010.10.025
Pubmed Absztrakt / Pubmed Teljes Szöveg / CrossRef Teljes Szöveg
20. B. I., Shen J., Timothy KW, Lehmann MH, Priori S., Robinson J. L., et al. A hosszú QT-szindróma génjeinek mutációinak spektruma. KVLQT1, HERG, SCN5A, KCNE1 és KCNE2. Keringés (2000) 102:1178-85. doi:10.1161 / 01.CIR.102.10.1178
Pubmed Absztrakt / Pubmed Teljes Szöveg / CrossRef Teljes Szöveg
21. Westenskow P, Splawski I, Timothy KW, Keating MT, Sanguinetti MC. Összetett mutációk: a súlyos long-QT szindróma gyakori oka. Keringés (2004) 109: 1834-41. doi:10.1161 / 01.CIR.0000125524.34234.13
Pubmed Absztrakt / Pubmed Teljes Szöveg / CrossRef Teljes Szöveg
22. Mullally J, Goldenberg I, Moss aj, Lopes CM, Ackerman MJ, Zareba W, et al. Életveszélyes kardiális események kockázata a hosszú QT-szindrómában és többszörös mutációban szenvedő betegek körében. Szívritmus (2013) 10: 378-82. doi: 10.1016 / j.2012.11.006
Pubmed Absztrakt / Pubmed Teljes Szöveg / CrossRef Teljes Szöveg
23. Napolitano C, Priori SG, Schwartz PJ, Bloise R, Ronchetti E, Nastoli J, et al. Genetikai tesztelés a hosszú QT-szindrómában: a genotipizálás hatékony megközelítésének kidolgozása és validálása a klinikai gyakorlatban. JAMA (2005) 294: 2975-80. doi: 10.1001 / jama.294.23.2975
Pubmed Absztrakt / Pubmed Teljes Szöveg / CrossRef Teljes Szöveg
24. Roden DM. Klinikai gyakorlat. Hosszú QT szindróma. N Engl J Med (2008) 358:169-76. doi: 10.1056 / NEJMcp0706513
CrossRef teljes szöveg
25. Schwartz PJ, Priori SG, Spazzolini C, Moss AJ, Vincent GM, Napolitano C, et al. Genotípus-fenotípus korreláció a long-QT szindrómában: az életveszélyes aritmiák génspecifikus kiváltói. Keringés (2001) 103: 89-95. doi:10.1161 / 01.CIR.103.1.89
CrossRef Teljes Szöveg
26. Ackerman MJ, tesztelő DJ, Porter CJ. Úszás, génspecifikus arrhythmogén kiváltó az öröklött hosszú QT szindrómához. Mayo Clin Proc (1999) 74:1088-94. doi:10.4065/74.11.1088
Pubmed absztrakt / Pubmed teljes szöveg / CrossRef teljes szöveg
27. Kapa S, tesztelő DJ, Salisbury BA, Harris-Kerr C, Pungliya MS, Alders M, et al. A hosszú QT-szindróma genetikai vizsgálata: a patogén mutációk megkülönböztetése a jóindulatú variánsoktól. Keringés (2009) 120: 1752-60. doi: 10.1161/CIRCULATIONAHA.109.863076
Pubmed Absztrakt / Pubmed Teljes Szöveg / CrossRef Teljes Szöveg
28. Moss AJ, Shimizu W, Wilde AA, Towbin JA, Zareba W, Robinson JL, et al. Az 1-es típusú long-QT szindróma klinikai vonatkozásai a kcnq1 gént érintő mutációk helye, kódolási típusa és biofizikai funkciója szerint. Keringés (2007) 115: 2481-9. doi: 10.1161/CIRCULATIONAHA.106.665406
Pubmed Absztrakt / Pubmed Teljes Szöveg / CrossRef Teljes Szöveg
29. Mint, Giudicessi JR, Tijsen AJ, Spanjaart AM, Reckman YJ, Klemens CA, et al. A kcnq1 által kódolt Kv7.1 káliumcsatorna 3′ nem lefordított régiójában található változatok allél-specifikus módon módosítják a betegség súlyosságát az 1-es típusú hosszú QT-szindrómában szenvedő betegeknél. Eur Szív J (2012) 33:714-23. doi: 10.1093 / eurheartj / ehr473
Pubmed absztrakt / Pubmed teljes szöveg / CrossRef teljes szöveg
30. Barsheshet A, Goldenberg I, O-Uchi J, Moss aj, Jons C, Shimizu W, et al. A kcnq1 csatorna citoplazmatikus hurkainak mutációi és az életveszélyes események kockázata: az 1. típusú long-QT szindróma mutáció-specifikus válaszának következményei a Alzheimer-blokkoló terápiára. Keringés (2012) 125: 1988-96. doi: 10.1161/CIRCULATIONAHA.111.048041
Pubmed Absztrakt / Pubmed Teljes Szöveg / CrossRef Teljes Szöveg
31. Schwartz PJ, Spazzolini C, Crotti L, Bathen J, Amlie JP, Timothy K, et al. A Jervell és a Lange-Nielsen szindróma: természettudomány, molekuláris alap és klinikai eredmény. Keringés (2006) 113: 783-90. doi: 10.1161/CIRCULATIONAHA.105.592899
Pubmed Absztrakt / Pubmed Teljes Szöveg / CrossRef Teljes Szöveg
32. Bhuiyan ZA, Wilde AA. A fül kevesebbel is megbirkózik, mint a szív. Circ Cardiovasc Genet (2013) 6:141-3. doi: 10.1161 / CIRCGENETICS.113.000143
CrossRef Teljes Szöveg
33. Wollnik B, Schroeder BC, Kubisch C, Esperer HD, Wieacker P, Jentsch TJ. Domináns és recesszív KVLQT1 k + csatorna mutációk patofiziológiai mechanizmusai örökletes szívritmuszavarokban. Hum Mol Genet (1997) 6:1943-9. doi:10.1093 / hmg / 6.11.1943
CrossRef Teljes Szöveg
34. Wilde AA, Jongbloed RJ, Doevendans PA, D .. D .. D .. Hauer RN, van Langen IM, et al. A hallási ingerek, mint az aritmiás események kiváltója, megkülönböztetik a HERG-rel kapcsolatos (LQTS2) betegeket a KVLQT1-rel kapcsolatos betegektől (LQTS1). J Am Coll Cardiol (1999) 33:327-32. doi: 10.1016 / S0735-1097(98)00578-6
CrossRef teljes szöveg
35. Moss aj, Zareba W, Kaufman ES, Gartman E, Peterson DR, Benhorin J, et al. Az aritmiás események fokozott kockázata hosszú QT-szindrómában mutációkkal az emberi éter-a-go-go-kapcsolódó gén káliumcsatorna pórusrégiójában. Keringés (2002) 105: 794-9. doi: 10.1161 / hc0702.105124
Pubmed absztrakt / Pubmed teljes szöveg / CrossRef teljes szöveg
36. Lupoglazoff JM, Denjoy I, gazember E, Fressart V, Simon F, Bozio a, et al. Hosszú QT-szindróma újszülötteknél: herg mutációkkal összefüggő vezetési zavarok és kcnq1 mutációkkal járó sinus bradycardia. J Am Coll Cardiol (2004) 43:826-30. doi: 10.1016 / j.jacc.2003.09.049
Pubmed Absztrakt / Pubmed Teljes Szöveg / CrossRef Teljes Szöveg
37. Csordás P, Bellocq C, Millat G, Piqueras E, Potet F, Schott JJ, et al. Torsades de pointes bonyolítja az atrioventrikuláris blokkot: bizonyíték a genetikai hajlamra. Szívritmus (2007) 4:170-4. doi: 10.1016 / j.2006.10.004
Pubmed Absztrakt / Pubmed Teljes Szöveg / CrossRef Teljes Szöveg
38. Hoorntje T, Alders M, van Tintelen P, Van der Lip K, Sreeram N, van der Wal A, et al. A HERG fehérje homozigóta korai csonkítása: az emberi HERG knockout. Keringés (1999) 100:1264-7. doi:10.1161 / 01.CIR.100.12.1264
Pubmed Absztrakt / Pubmed Teljes Szöveg / CrossRef Teljes Szöveg
39. Piippo K, Laitinen P, hattyú H, Toivonen L, Viitasalo M, Pasternack M, et al. A HERG káliumcsatorna-mutáció homozigozitása a hosszú QT-szindróma súlyos formáját okozza: egy látszólagos alapító mutáció azonosítása a Finnekben. J Am Coll Cardiol (2000) 35:1919-25. doi: 10.1016 / S0735-1097(00)00636-7
Pubmed absztrakt / Pubmed teljes szöveg / CrossRef teljes szöveg
40. Johnson WH Jr, Yang P, Yang T, Lau év, Mostella BA, Wolff DJ, et al. A súlyos újszülöttkori hosszú QT szindrómát okozó homozigóta HERG mutáció klinikai, genetikai és biofizikai jellemzése. Pediatr Res (2003) 53:744-8. doi:10.1203 / 01.PDR.0000059750.17002.B6
Pubmed Absztrakt / Pubmed Teljes Szöveg / CrossRef Teljes Szöveg
41. Ackerman MJ, tesztelő DJ, Jones GS, will ML, Burrow CR, Curran nekem. Etnikai különbségek a szív káliumcsatorna variánsaiban: a hirtelen szívhalálra való genetikai fogékonyság és a veleszületett hosszú QT-szindróma genetikai vizsgálata. Mayo Clin Proc (2003) 78:1479-87. doi:10.4065/78.12.1479
Pubmed absztrakt / Pubmed teljes szöveg / CrossRef teljes szöveg
42. Crotti L, Lundquist AL, Insolia R, Pedrazzini M, Ferrandi C, De Ferrari GM, et al. A KCNH2-K897T a látens veleszületett hosszú QT szindróma genetikai módosítója. Keringés (2005) 112:1251-8. doi: 10.1161/CIRCULATIONAHA.105.549071
Pubmed Absztrakt / Pubmed Teljes Szöveg / CrossRef Teljes Szöveg
43. Nof E, Cordeiro JM, P vállalkozók GJ, Scornik FS, Calloe K, szerelem B, et al. A közös egyetlen nukleotid polimorfizmus súlyosbíthatja a long-QT típusú 2 szindrómát, amely hirtelen csecsemőhalálhoz vezet. Circ Cardiovasc Genet (2010) 3:199-206. doi: 10.1161 / CIRCGENETICS.109.898569
Pubmed Absztrakt / Pubmed Teljes Szöveg / CrossRef Teljes Szöveg
44. Bezzina C, Veldkamp MW, van den Berg MP, Postma AV, Bástya MB, Viersma JW, et al. Egyetlen Na (+) csatorna mutáció, amely mind a long-QT, mind a Brugada szindrómákat okozza. Circ Res (1999) 85:1206-13. doi:10.1161 / 01.RES.85.12.1206
Pubmed absztrakt / Pubmed teljes szöveg / CrossRef teljes szöveg
45. Kyndt F, Probst V, Potet F, Demolombe S, Chevallier JC, Baro I, et al. Új SCN5A mutáció, amely izolált szívvezetési hibához vagy Brugada-szindrómához vezet egy nagy francia családban. Keringés (2001) 104:3081-6. doi: 10.1161 / hc5001.100834
Pubmed absztrakt / Pubmed teljes szöveg / CrossRef teljes szöveg
46. Makita N, Behr E, Shimizu W, Horie M, Sunami A, Crotti L, et al. Az SCN5A E1784K mutációja a 3-as típusú hosszú QT szindróma vegyes klinikai fenotípusához kapcsolódik. J Clin Invest (2008) 118:2219-29. doi: 10.1172 / JCI34057
Pubmed absztrakt / Pubmed teljes szöveg / CrossRef teljes szöveg
47. Keller DI, Acharfi S, Delacr Enterprises e, Benammar N, Rotter M, Pfammatter JP, et al. Új mutáció az SCN5A-ban, delQKP 1507-1509, hosszú QT-szindrómát okozva: a Q1507 maradék szerepe a nátriumcsatorna inaktiválásában. J Mol Cell Cardiol (2003) 35:1513-21. doi:10.1016 / j.2003.08.007
Pubmed Absztrakt / Pubmed Teljes Szöveg / CrossRef Teljes Szöveg
48. Priori SG, Schwartz PJ, Napolitano C, Bloise R, Ronchetti E, Grillo M, et al. Kockázati rétegződés a hosszú QT szindrómában. N Engl J Med (2003) 348:1866-74. doi:10.1056 / NEJMoa022147
Pubmed absztrakt / Pubmed teljes szöveg / CrossRef teljes szöveg
49. Lupoglazoff JM, Cheav T, Baroudi G, Berthet M, Denjoy I, Cauchemez B, et al. Homozigóta SCN5A mutáció hosszú QT szindrómában funkcionális két-egy atrioventrikuláris blokkkal. Circ Res (2001) 89:E16–21. doi:10.1161 / hh1401. 095087
CrossRef teljes szöveg
50. Shi R, Zhang Y, Yang C, Huang C, Zhou X, Qiang H, et al. A delqkp 1507-1509 kardiális nátriumcsatorna mutáció az LQT3 bővülő fenotípusos spektrumával, vezetési zavarral, dilatált kardiomiopátiával és a fiatalok hirtelen halálának magas előfordulásával jár. Europace (2008) 10:1329-35. doi: 10.1093 / europace / eun202
Pubmed absztrakt / Pubmed teljes szöveg / CrossRef teljes szöveg
51. Zumhagen S, Veldkamp MW, Stallmeyer B, Baartscheer A, Eckardt L, Paul M, et al. A cardialis nátriumcsatorna gén scn5a heterozigóta deléciós mutációja veszteség-és funkciógyarapodási jellemzőkkel izolált vezetési betegségként nyilvánul meg, Brugada vagy long QT szindróma jelei nélkül. PLoS egy (2013) 8:e67963. doi: 10.1371 / folyóirat.pone.0067963
Pubmed Absztrakt / Pubmed Teljes Szöveg / CrossRef Teljes Szöveg
52. Növény LD, Bowers PN, Liu Q, Morgan T, Zhang T, állami MW, et al. Gyakori szív-nátriumcsatorna-variáns, amely hirtelen csecsemőhalálhoz kapcsolódik afroamerikaiaknál, SCN5A S1103Y.J Clin Invest (2006) 116:430-5. doi:10.1172 / JCI25618
Pubmed absztrakt / Pubmed teljes szöveg / CrossRef teljes szöveg
53. Mohler PJ, Schott JJ, Gramolini AO, Dilly KW, Guatimosim S, duBell WH, et al. Az Ankyrin-B mutáció 4-es típusú long-QT szívritmuszavart és hirtelen szívhalált okoz. Természet (2003) 421:634-9. doi: 10.1038 / nature01335
Pubmed absztrakt / Pubmed teljes szöveg / CrossRef teljes szöveg
54. Schott JJ, Charpentier F, Peltier S, Foley P, Drouin E, Bouhour JB, et al. A hosszú QT-szindróma génjének feltérképezése a 4q25-27 kromoszómára. Am J Hum Genet (1995) 57:1114-22.
Pubmed Absztrakt / Pubmed Teljes Szöveg
55. Barhanin J, Lesage F, Guillemare E, Fink M, Lazdunski M, Romey G. K(V)LQT1 és lsK (minK) fehérjék társulnak az I(ks) kardiális káliumáram kialakításához. Természet (1996) 384:78-80. doi: 10.1038 / 384078a0
Pubmed absztrakt / Pubmed teljes szöveg / CrossRef teljes szöveg
56. Sanguinetti MC, Curran ME, Zou A, Shen J, Spector PS, Atkinson DL, et al. K(V)LQT1 és minK (ISK) fehérjék együttes összeszerelése a szív I(Ks) káliumcsatorna kialakításához. Természet (1996) 384:80-3. doi:10.1038/384080a0
Pubmed absztrakt / Pubmed teljes szöveg / CrossRef teljes szöveg
57. Schulze-Bahr E, Wang Q, Wedekind H, Haverkamp W, Chen Q, Sun Y, et al. A KCNE1 mutációk Jervell és Lange-Nielsen szindrómát okoznak. Nat Genet (1997) 17:267-8. doi: 10.1038 / ng1197-267
CrossRef teljes szöveg
58. Duggal P, Vesely Úr, Wattanasirichaigoon D, Villafane J, Kaushik V, Beggs AH. Az IKs gén mutációja mind a Jervell, mind a Lange-Nielsen, valamint a long-QT szindróma Romano-Ward formáival összefüggésben. Keringés (1998) 97:142-6. doi:10.1186/1471-2350-9-24
Pubmed Absztrakt / Pubmed Teljes Szöveg / CrossRef Teljes Szöveg
59. Nishio Y, Makiyama T, Itoh H, Sakaguchi T, Ohno S, Gong YZ, et al. A D85N, a kcne1 polimorfizmus, egy betegséget okozó génvariáns hosszú QT-szindrómában. J Am Coll Cardiol (2009) 54:812-9. doi: 10.1016 / j.jacc.2009.06.005
Pubmed Absztrakt / Pubmed Teljes Szöveg / CrossRef Teljes Szöveg
60. Paulussen AD, Gilissen RA, Armstrong M, Doevendans PA, Verhasselt P, Smeets HJ, et al. A kcnq1, KCNH2, SCN5A, KCNE1 és KCNE2 genetikai variációi gyógyszer által indukált hosszú QT szindrómás betegeknél. Mol Med (2004) 82:182-8. doi: 10.1007 / s00109-003-0522-Z
Pubmed absztrakt / Pubmed teljes szöveg / CrossRef teljes szöveg
61. Abbott GW, Sesti F, Splawski I, Buck ME, Lehmann MH, Timothy KW és mtsai. A MiRP1 IKr káliumcsatornákat képez a HERG-vel, és szívritmuszavarhoz kapcsolódik. Cell (1999) 97:175-87. doi: 10.1016 / S0092-8674 (00) 80728-X
Pubmed absztrakt / Pubmed teljes szöveg / CrossRef teljes szöveg
62. Gordon E, Panaghie G, Deng L, Bee KJ, Roepke TK, Krogh-Madsen T, et al. Kcne2 mutáció szívritmuszavarban szenvedő betegben, melyet hallási ingerek és a szérum elektrolit-egyensúlyzavar vált ki. Cardiovasc Res (2008) 77:98-106. doi: 10.1093 / cvr/cvm030
Pubmed absztrakt / Pubmed teljes szöveg / CrossRef teljes szöveg
63. Plasting NM, Tawil R, Tristani-Firouzi M, Can 6N S, Bendahhou S, Tsunoda A, et al. A Kir2.1 mutációi az Andersen-szindróma fejlődési és epizodikus elektromos fenotípusait okozzák. Cell (2001) 105:511-9. doi:10.1016 / S0092-8674(01)00342-7
Pubmed Absztrakt / Pubmed Teljes Szöveg / CrossRef Teljes Szöveg
64. Kubo Y, Baldwin TJ, Jan YN, Jan LY. Elsődleges szerkezete és funkcionális kifejezése egy egér befelé egyenirányító kálium-csatorna. Természet (1993) 362:127-33. doi: 10.1038 / 362127a0
Pubmed absztrakt / Pubmed teljes szöveg / CrossRef teljes szöveg
65. Raab-Graham KF, Radeke CM, Vandenberg ca. Molekuláris klónozás és kifejezése egy emberi szív befelé egyenirányító kálium csatorna. Neuroreport (1994) 5:2501-5. doi:10.1097/00001756-199412000-00024
Pubmed Absztrakt / Pubmed Teljes Szöveg / CrossRef Teljes Szöveg
66. Splawski I, Timothy KW, Sharpe LM, Decher N, Kumar P, Bloise R és mtsai. Ca (V) 1.2 a kalciumcsatorna diszfunkció multisystem rendellenességet okoz, beleértve az aritmiát és az autizmust. Cell (2004) 119:19-31. doi:10.1016 / j. cella.2004.09.011
Pubmed Absztrakt / Pubmed Teljes Szöveg / CrossRef Teljes Szöveg
67. Splawski I, Timothy KW, Decher N, Kumar P, Sachse FB, Beggs AH, et al. Súlyos aritmia rendellenesség, amelyet szív L-típusú kalciumcsatorna mutációk okoznak. Proc Natl Acad Sci U S A (2005) 102:8089-96. doi: 10.1073/pnas.0502506102
Pubmed Absztrakt / Pubmed Teljes Szöveg / CrossRef Teljes Szöveg
68. Gillis J, Burasnyikov E, Antzelevitch C, Blaser S, bruttó G, Turner L, et al. Hosszú QT, syndactyly, ízületi kontraktúrák, stroke és új cacna1c mutáció: a Timothy-szindróma spektrumának bővítése. Am J Med Genet A (2012) 158A:182-7. doi:10.1002 / ajmg.a.34355
Pubmed Absztrakt / Pubmed Teljes Szöveg / CrossRef Teljes Szöveg
69. Dufendach KA, Giudicessi JR, Boczek NJ, Ackerman MJ. Az anyai mozaikosság megzavarja az 1. típusú Timothy-szindróma újszülöttkori diagnózisát. Gyermekgyógyászat (2013) 131:e1991–5. doi: 10.1542 / peds.2012-2941
Pubmed Absztrakt / Pubmed Teljes Szöveg / CrossRef Teljes Szöveg
70. Vatta M, Ackerman MJ, Ye B, Makielski JC, Ughanze EE, Taylor EW, et al. A mutáns caveolin-3 tartós késői nátriumáramot indukál, és hosszú QT szindrómával jár. Forgalom (2006) 114:2104–12. doi: 10.1161/CIRCULATIONAHA.106.635268
Pubmed Absztrakt / Pubmed Teljes Szöveg / CrossRef Teljes Szöveg
71. Cronk LB, ti B, Kaku T, tesztelő DJ, Vatta M, Makielski JC, et al. A hirtelen csecsemőhalál szindróma új mechanizmusa: a caveolin-3 mutációi miatt másodlagos tartós késői nátriumáram. Szívritmus (2007) 4:161-6. doi: 10.1016 / j.2006.11.030
Pubmed Absztrakt / Pubmed Teljes Szöveg / CrossRef Teljes Szöveg
72. Engelman JA, Zhang X, Galbiati F, Volonte D, Sotgia F, Pestell RG, et al. A caveolin géncsalád molekuláris genetikája: következményei az emberi rákokra, a cukorbetegségre, az Alzheimer-kórra és az izomdisztrófiára. Am J Hum Genet (1998) 63:1578-87. doi:10.1086/302172
CrossRef teljes szöveg
73. Balijepalli RC, Foell JD, Hall DD, pokol JW, Kamp TJ. A szív L-típusú Ca (2+) csatornáinak lokalizálása caveoláris makromolekuláris jelátviteli komplexhez szükséges a béta(2)-adrenerg szabályozáshoz. Proc Natl Acad Sci U S A (2006) 103:7500-5. doi: 10.1073/pnas.0503465103
Pubmed Absztrakt / Pubmed Teljes Szöveg / CrossRef Teljes Szöveg
74. Medeiros-Domingo a, Kaku T, tesztelő DJ, Iturralde-Torres P, Itty A, ti B, et al. SCN4B-kódolt nátriumcsatorna béta4 alegység veleszületett hosszú QT szindrómában. Keringés (2007) 116: 134-42. doi: 10.1161/CIRCULATIONAHA.106.659086
Pubmed Absztrakt / Pubmed Teljes Szöveg / CrossRef Teljes Szöveg
75. Chen L, Marquardt ML, tesztelő DJ, Sampson KJ, Ackerman MJ, Kass RS. Az A-kináz-rögzítő fehérje mutációja hosszú QT-szindrómát okoz. Proc Natl Acad Sci U S A (2007) 104:20990-5. doi: 10.1073/pnas.0710527105
Pubmed Absztrakt / Pubmed Teljes Szöveg / CrossRef Teljes Szöveg
76. Wu G, Ai T, Kim JJ, Mohapatra B, Xi Y, Li Z, et al. Alfa-1-syntrophin mutáció és a hosszú QT-szindróma: a nátriumcsatorna-zavar betegsége. Circ Arrhythm Elektrofiziol (2008) 1:193-201. doi: 10.1161 / CIRCEP.108.769224
Pubmed Absztrakt / Pubmed Teljes Szöveg / CrossRef Teljes Szöveg
77. Yang Y, Yang Y, Liang B, Liu J, Li J, Grunnet M, et al. Kir3 azonosítása.4 mutáció veleszületett hosszú QT-szindrómában. Am J Hum Genet (2010) 86:872-80. doi: 10.1016 / j.2010.04.017
Pubmed Absztrakt / Pubmed Teljes Szöveg / CrossRef Teljes Szöveg
78. Roden DM. A proarrhythmia mechanizmusai és kezelése. Am J Cardiol (1998) 82:49I–57I. doi: 10.1016 / S0002-9149(98) 00472-X
CrossRef teljes szöveg
79. Mitcheson JS, Chen J, Lin M, Culberson C, Sanguinetti MC. A gyógyszer által kiváltott hosszú QT-szindróma szerkezeti alapja. Proc Natl Acad Sci U S A (2000) 97:12329-33. doi: 10.1073/pnas.210244497
CrossRef Teljes Szöveg
80. Kannankeril PJ, Roden DM. Kábítószer-indukált hosszú QT és torsade de pointes: legutóbbi előrelépések. Curr Opin Cardiol (2007) 22:39-43. doi:10.1097 / HCO.0b013e32801129eb
Pubmed absztrakt / Pubmed teljes szöveg / CrossRef teljes szöveg
81. V. C., Verkerk AO, Blom MT, Klemens CA, Langendijk PN, van Ginneken AC, et al. A lassú késleltetett egyenirányító káliumáram-blokád fontos szerepet játszik a gyógyszer által kiváltott hosszú QT-szindrómában. Circ Arrhythm Elektrofiziol (2013) 6:1002-9. doi: 10.1161 / CIRCEP.113.000239
Pubmed Absztrakt / Pubmed Teljes Szöveg / CrossRef Teljes Szöveg
82. Sesti F, Abbott GW, Wei J, Murray KT, Saksena S, Schwartz PJ, et al. Az antibiotikum által kiváltott szívritmuszavarhoz kapcsolódó gyakori polimorfizmus. Proc Natl Acad Sci U S A (2000) 97:10613-8. doi: 10.1073/pnas.180223197
Pubmed Absztrakt / Pubmed Teljes Szöveg / CrossRef Teljes Szöveg
83. K kb s, Crawford DC, bűnös MF, Behr er, kannankeril pj, Wilde aa, et al. Egy nagy jelölt génfelmérés azonosítja a KCNE1 D85N polimorfizmus a gyógyszer által kiváltott torsades de pointes lehetséges modulátoraként. Circ Cardiovasc Genet (2012) 5:91-9. doi: 10.1161 / CIRCGENETICS.111.960930
Pubmed Absztrakt / Pubmed Teljes Szöveg / CrossRef Teljes Szöveg
84. Nakamura K, Katayama Y, Kusano KF, Haraoka K, Tani Y, Nagase s, et al. Anti-KCNH2 antitest által kiváltott hosszú QT szindróma: a hosszú QT szindróma új szerzett formája. J Am Coll Cardiol (2007) 50:1808-9. doi: 10.1016 / j.jacc.2007.07.037
CrossRef Teljes Szöveg
85. Moss AJ, Zareba W, Benhorin J, Locati EH, Hall WJ, Robinson JL, et al. EKG T-hullám minták az örökletes hosszú QT-szindróma genetikailag különböző formáiban. Keringés (1995) 92:2929-34. doi:10.1161 / 01.CIR.92.10.2929
Pubmed Absztrakt / Pubmed Teljes Szöveg / CrossRef Teljes Szöveg
86. J. L., Timothy KW, Vincent GM, Lehmann MH, Fox J., Giuli LC, et al. Az ST-T-hullám minták spektruma és a repolarizációs paraméterek veleszületett hosszú QT szindrómában: az EKG-eredmények azonosítják a genotípusokat. Keringés (2000) 102: 2849-55. doi:10.1161 / 01.CIR.102.23.2849
Pubmed Absztrakt / Pubmed Teljes Szöveg / CrossRef Teljes Szöveg
87. Tan HL, Bardai A, Shimizu W, Moss AJ, Schulze-Bahr E, Noda T, et al. Az aritmiák genotípus-specifikus kialakulása veleszületett long-QT szindrómában: lehetséges terápiás következmények. Keringés (2006) 114: 2096-103. doi: 10.1161/CIRCULATIONAHA.106.642694
Pubmed Absztrakt / Pubmed Teljes Szöveg / CrossRef Teljes Szöveg
88. Locati EH, Zareba W, Moss AJ, Schwartz PJ, Vincent GM, Lehmann MH, et al. A veleszületett long – QT szindrómában szenvedő betegek klinikai megnyilvánulásainak életkorral és nemekkel kapcsolatos különbségei: a nemzetközi LQTS nyilvántartás megállapításai. Keringés (1998) 97:2237-44. doi:10.1161 / 01.CIR.97.22.2237
Pubmed Absztrakt / Pubmed Teljes Szöveg / CrossRef Teljes Szöveg
89. Zareba W, Moss AJ, Schwartz PJ, Vincent GM, Robinson JL, Priori SG, et al. A genotípus hatása a long-QT szindróma klinikai lefolyására. Nemzetközi hosszú QT szindróma nyilvántartási kutatócsoport. N Engl J Med (1998) 339:960-5. doi:10.1056 / NEJM199810013391404
Pubmed absztrakt / Pubmed teljes szöveg / CrossRef teljes szöveg
90. Goldenberg I, Moss AJ, Bradley J, Polonsky S, Peterson DR, McNitt S, et al. Hosszú QT-szindróma 40 éves kor után. Keringés (2008) 117: 2192-201. doi: 10.1161/CIRCULATIONAHA.107.729368
Pubmed Absztrakt / Pubmed Teljes Szöveg / CrossRef Teljes Szöveg
91. Zareba W, Moss AJ, Locati EH, Lehmann MH, Peterson DR, Hall WJ, et al. Szindróma Nyilvántartás. Az életkor és a nem moduláló hatása a hosszú QT-szindróma klinikai lefolyására genotípus szerint. J Am Coll Cardiol (2003) 42:103-9. doi: 10.1016 / S0735-1097(03)00554-0
Pubmed absztrakt / Pubmed teljes szöveg / CrossRef teljes szöveg
92. Seth R, Moss AJ, Mcnitt S, Zareba W, Andrews ML, Qi M, et al. Hosszú QT szindróma és terhesség. J Am Coll Cardiol (2007) 49:1092-8. doi: 10.1016 / j.jacc.2006.09.054
Pubmed Absztrakt / Pubmed Teljes Szöveg / CrossRef Teljes Szöveg
93. Schwartz PJ, Priori SG, Dumaine R, Napolitano C, Antzelevitch C, Stramba-Badiale M, et al. Molekuláris kapcsolat a hirtelen csecsemőhalál szindróma és a long-QT szindróma között. N Engl J Med (2000) 343:262-7. doi: 10.1056 / NEJM200007273430405
CrossRef teljes szöveg
94. Schwartz PJ, Priori SG, Bloise R, Napolitano C, Ronchetti E, Piccinini A, et al. Molekuláris diagnózis hirtelen csecsemőhalál szindrómában szenvedő gyermeknél. Lancet (2001) 358:1342-3. doi: 10.1016 / S0140-6736(01)06450-9
Pubmed absztrakt / Pubmed teljes szöveg / CrossRef teljes szöveg
95. Christiansen M, T), Larsen LA, Andersen PS, Simonsen H, Oyen N, et al. Mutációk a HERG K + – ion csatornában: új kapcsolat a hosszú QT szindróma és a hirtelen csecsemőhalál szindróma között. Am J Cardiol (2005) 95:433-4. doi: 10.1016 / j.amjcard.2004.09.054
Pubmed Absztrakt / Pubmed Teljes Szöveg / CrossRef Teljes Szöveg
96. Tesztelő DJ, Ackerman MJ. Postmortem hosszú QT szindróma genetikai vizsgálat hirtelen megmagyarázhatatlan halál a fiatalok. J Am Coll Cardiol (2007) 49:240-6. doi: 10.1016 / j.jacc.2006.10.010
Pubmed Absztrakt / Pubmed Teljes Szöveg / CrossRef Teljes Szöveg
97. Hofman N, Tan HL, Clur SA, Alders M, van Langen IM, Wilde AA. Az öröklött szívbetegség hozzájárulása a gyermekkori hirtelen szívhalálhoz. Gyermekgyógyászat (2007) 120:e967–73. doi: 10.1542 / peds.2006-3751
Pubmed Absztrakt / Pubmed Teljes Szöveg / CrossRef Teljes Szöveg
98. Wedekind H, Smits JP, Schulze-Bahr E, Arnold R, Veldkamp MW, Bajanowski T, et al. De novo mutáció az SCN5A génben, amely a hirtelen csecsemőhalál korai megjelenésével jár. Keringés (2001) 104: 1158-64. doi: 10.1161 / hc3501. 095361
CrossRef teljes szöveg
99. Wang DW, Desai RR, Crotti L, Arnestad M, Insolia R, Pedrazzini M, et al. Szív nátriumcsatorna diszfunkció hirtelen csecsemőhalál szindrómában. Keringés (2007) 115: 368-76. doi: 10.1161/CIRCULATIONAHA.106.646513
Pubmed Absztrakt / Pubmed Teljes Szöveg / CrossRef Teljes Szöveg
100. Van Norstrand DW, Valdivia CR, tesztelő DJ, Ueda K, London B, Makielski JC, et al. Új glicerin-3-foszfát-dehidrogenáz 1-szerű gén (GPD1-L) mutációk molekuláris és funkcionális jellemzése hirtelen csecsemőhalál szindrómában. Keringés (2007) 116: 2253-9. doi:10.1161 / CIRCULATIONAHA.107.704627
Pubmed Absztrakt / Pubmed Teljes Szöveg / CrossRef Teljes Szöveg
101. Tan BH, Pundi KN, van Norstrand DW, Valdivia CR, tesztelő DJ, Medeiros-Domingo a, et al. Hirtelen csecsemőhalál szindrómával összefüggő mutációk a nátriumcsatorna béta alegységeiben. Szívritmus (2010) 7:771-8. doi: 10.1016 / j.2010.01.032
Pubmed Absztrakt / Pubmed Teljes Szöveg / CrossRef Teljes Szöveg
102. Miller TE, Estrella E, Myerburg RJ, Garcia de Viera J, Moreno N, Rusconi P, et al. Visszatérő harmadik trimeszter magzati veszteség és anyai mozaikosság hosszú QT szindróma esetén. Keringés (2004) 109: 3029-34. doi:10.1161 / 01.CIR.0000130666.81539.9 E
Pubmed Absztrakt / Pubmed Teljes Szöveg / CrossRef Teljes Szöveg
103. Chockalingam P, Crotti L, Girardengo G, Johnson JN, Harris KM, van der Heijden JF, et al. Nem minden béta-blokkoló azonos az 1-es és 2-es típusú hosszú QT-szindróma kezelésében: a metoprolol alatt bekövetkező események nagyobb ismétlődése. J Am Coll Cardiol (2012) 60:2092-6. doi: 10.1016 / j.jacc.2012.07.046
Pubmed Absztrakt / Pubmed Teljes Szöveg / CrossRef Teljes Szöveg
104. Schwartz PJ, Priori SG, Cerrone M, Spazzolini C, Odero A, Napolitano C, et al. Bal szív szimpatikus denerváció a hosszú QT-szindróma által érintett magas kockázatú betegek kezelésében. Keringés (2004) 109: 1826-33. doi:10.1161 / 01.CIR.0000125523.14403.1 E
Pubmed Absztrakt / Pubmed Teljes Szöveg / CrossRef Teljes Szöveg
105. Singh B, al Shahwan SA, Habbab MA, al Deeb SM, Biary N. idiopátiás hosszú QT szindróma: a helyes kérdés feltevése. Lancet (1993) 341:741. doi:10.1016/0140-6736(93)90501-7
CrossRef teljes szöveg
106. Gorgels AP, Al Fadley F, Zaman L, Kantoch MJ, Al Halees Z. a hosszú QT-szindróma károsodott atrioventrikuláris vezetéssel: rosszindulatú változat csecsemőknél. J Cardiovasc Elektrofiziol (1998) 9:1225-32. doi: 10.1111 / j. 1540-8167.1998.tb00096.x
Pubmed absztrakt / Pubmed teljes szöveg / CrossRef teljes szöveg
107. Kantoch MJ, Qurashi MM, Bulbul ZR, Gorgels AP. Egy újszülött komplex veleszületett szívbetegség, atrioventricularis blokk, torsade de pointes kamrai tachycardia. Ingerlő Clin Elektrofiziol (1998) 21: 2664-7. doi: 10.1111 / j. 1540-8159.1998.tb00043.x
CrossRef teljes szöveg
108. El-Hazmi MA, al-Swailem AR, Warsy AS, al-Swailem AM, Sulaimani R, al-Meshari AA. Rokonság a szaúd-arábiai lakosság körében. J Med Genet (1995) 32:623-6. doi: 10.1136 / jmg.32.8.623
CrossRef Teljes Szöveg
109. El Mouzan MI, Al SALLOUM AA, Al Herbish AS, Qurachi MM, Al Omar AA. Rokonság és súlyos genetikai rendellenességek Szaúdi gyermekeknél: közösségi alapú keresztmetszeti vizsgálat. Ann Szaúdi Med (2008) 28:169-73. doi:10.4103/0256-4947.51726
Pubmed absztrakt / Pubmed teljes szöveg / CrossRef teljes szöveg
110. Medeiros-Domingo A, Bhuiyan ZA, tesztelő DJ, Hofman N, Bikker H, van Tintelen JP, et al. A RYR2-kódolt ryanodin receptor / kalcium felszabadulási csatorna korábban diagnosztizált betegeknél katekolaminerg polimorf kamrai tachycardia vagy genotípus negatív, testmozgás által kiváltott hosszú QT szindróma: átfogó nyílt olvasási keret mutációs elemzés. J Am Coll Cardiol (2009) 54:2065-74. doi: 10.1016 / j.jacc.2009.08.022
Pubmed Absztrakt / Pubmed Teljes Szöveg / CrossRef Teljes Szöveg
111. Al-Aama JY, Al-Ghamdi S, Bdier AY, Wilde AA, Bhuiyan ZA. A KCNQ1 gén de novo mutációja okozza a Jervell és a Lange-Nielsen szindrómát. Clin Genet (2013). doi:10.1111 / cge.12300
Pubmed Absztrakt / Pubmed Teljes Szöveg / CrossRef Teljes Szöveg
112. Tóth M., Tóth M., Tóth M., Tóth A., Bégg Á., et al. Az SCN5A nátriumcsatorna változata, amely szerepet játszik a szívritmuszavar kockázatában. Tudomány (2002) 297:1333-6. doi: 10.1126 / tudomány.1073569
CrossRef Teljes Szöveg
113. Wilde AA, Bezzina CR. A szívritmuszavarok genetikája. Szív (2005) 91:1352-8.