Mukopoliszacharidózisok: korai diagnosztikai jelek csecsemőknél és gyermekeknél
a Mukopoliszacharidózisok (MPS) klinikailag heterogén betegségek csoportja, amelyeket a glikozaminoglikánok (Gag) lebontásához szükséges lizoszomális enzimek hiánya okoz.
az MPS-t progresszív és szisztémás klinikai tünetek jellemzik. Biokémiai és genetikai heterogenitásuk ellenére a különböző típusok különböző kombinációkban osztják meg a legfontosabb klinikai jellemzőket, beleértve a merevséggel járó ízületi és csontváz diszpláziát (kivéve az MPS IV-et, ahol lazaság van) és fájdalmat, durva arcvonásokat, szaruhártya-elhomályosodást, inguinalis vagy hasi sérveket, visszatérő felső légúti fertőzések, szívbillentyű-betegség, carpalis alagút szindróma és változó neurológiai érintettség. Ezek a jellemzők általában az élet első hónapjaiban jelennek meg a súlyos formáknál és a korai gyermekkorban a gyengített formáknál, de gyakran alábecsülik őket, és általában csak akkor veszik figyelembe őket, ha egyértelműen nyilvánvalóak.
ezeknek a rendellenességeknek a ritkasága és a klinikai megjelenés változékonysága gyakran diagnosztikai késleltetéshez vezet; ez a súlyos formák esetében hónapoktól évekig terjedhet, az attenuáltaknál (lásd Rigoldi et al. ebben a kiegészítésben). Mindazonáltal a gyors előrehaladás és a súlyos prezentációk sürgős beavatkozásának szükségessége miatt a diagnózis több hónapos késése katasztrofális eredményeket hozhat a beteg jövőbeli egészségére nézve.
célunk, hogy ebben a felülvizsgálatban hangsúlyozzuk a súlyos formák nagyon korai jeleit, amelyeknek figyelmeztetniük kell a gyermekorvost az első megjelenésre.
milyen jelek / tünetek várhatók a súlyos MPS-ben szenvedő gyermekeknél?
az MPS különböző formáira utaló tüneteket és jeleket az 1.táblázat mutatja életkor szerint. Az élet első 6 hónapjában lágyéksérv, rendellenesen gyakori légúti fertőzések, otitis és organomegalia figyelhető meg MPS betegeknél, különösen az MPS I – ben (Hurler-szindróma), amelynek a legsúlyosabb súlyos megjelenése van. Továbbá, 6-12 hónap, ezek a csecsemők nagyon gyakran alakulnak ki gibbus (toraco-lumbális kyphosis), szívzörej, köldöksérv, enyhe hypotonia, növekedési késleltetés . Lehet, hogy tipikus durva arckifejezésük is van (ábra. 1). Az MPS II, VI és VII hasonló korai fenotípussal rendelkezhet. Az MPS IV betegek korai csontváz-fenotípussal rendelkeznek, csípő diszplázia megjelenésével az élet első hónapjaiban, a galamb mellkasával (pectus carinatum) az első 12-18 hónapban. Az MPS II csecsemők hasonló érintettséggel, de később jelentkeznek. A második életévben az MPS I Hurler-szindrómás csecsemőknél kognitív késés alakul ki, míg az MPS II beteget csak a túlnövekedés, a gyakori légúti fertőzések és a többszörös műtétek miatt lehet azonosítani ; a tipikus arcvonások csak később jelennek meg. Néhány MPS II betegnél hiperaktivitás is kialakul 18-24 hónap között. Az élet harmadik évében a hiperaktivitás és a kognitív késleltetés könnyen felismerhető az MPS II és az MPS III.
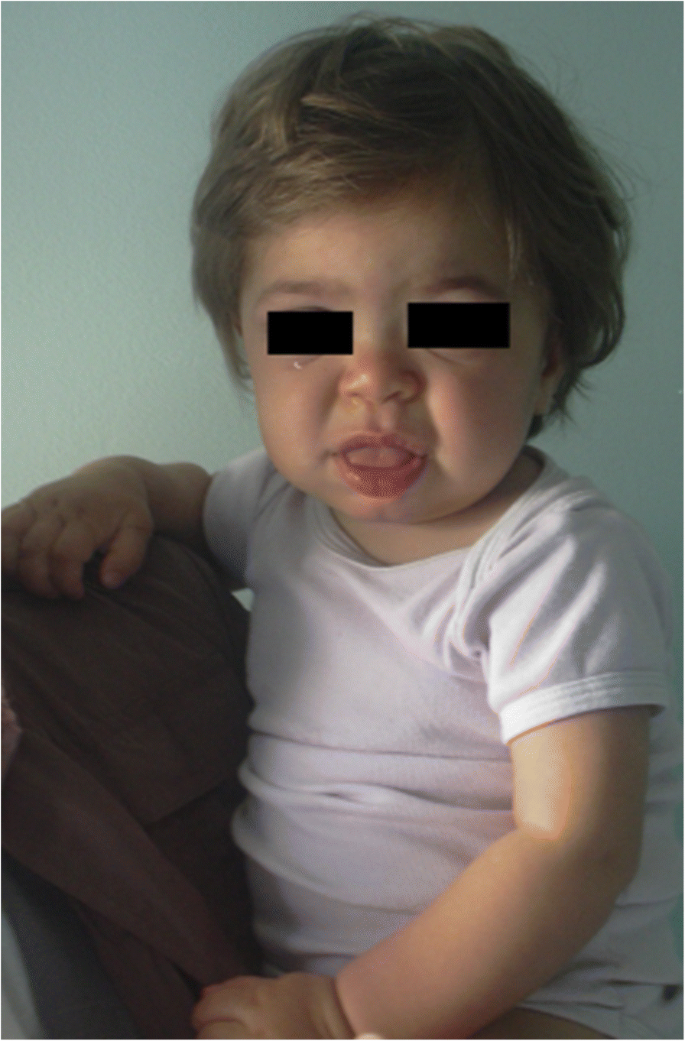
MPS IH 1 éves betegben. Durva facies: lapos orrhíd, macroglossia, frontális főnök
összefoglalva, a csontváz megnyilvánulásai tipikusak, néha koraérettek. A durva facies és a kognitív késleltetés nem korai kiemelkedő jelek.
melyek a különböző típusú MPS különböző bemutatói?
szomatikus és kognitív érintettséggel rendelkező MPS
I. típusú mukopoliszacharidózis (MPS I; OMIM #252800) (ábra. 1., 2., 3., 4. és 5.) a lizoszomális hidroláz hiánya okozza, amely két Gag multiszisztémás felhalmozódásához vezet: dermatán-szulfát (DS) és heparán-szulfát (HS). Súlyossága alapján az MPS I hagyományosan három klinikai fenotípusra oszlik; ezek a formák azonban a betegség súlyosságának folytonosságát képviselik. A Hurler-szindróma, a legsúlyosabb forma, általában az első életévben jelentkezik. Az érintett gyermekek gyorsan jelentős kognitív károsodást és szomatikus betegséget fejlesztenek ki több szervrendszerben, ami kezelés hiányában az első évtizedben halálhoz vezet. Az MPS I gyengített formáit, amelyeket Hurler–Scheie szindrómának és Scheie szindrómának neveznek, a tünetek későbbi megjelenése, a hosszabb várható élettartam és a központi idegrendszer (CNS) enyhe vagy egyáltalán nem érintettsége jellemzi . Az öröklés autoszomális recesszív, az esetek legalább 50% – ában a fenotípus előrejelzése a genotípus alapján lehetséges, míg a fennmaradó 50% – ban új mutációkat észlelnek . A prevalencia körülbelül 1: 100 000 élveszületés .

MPS IH. Gibbus egy 9 hónapos betegben. b gerinc röntgen a gerinc egy 3 éves beteg. c T2-súlyozott mágneses rezonancia kép a gerincről, amely jelentős kyphosisot és gerinccsatorna stenosist mutat

MPS IH. Röntgen bizonyíték dysostosis multiplex. a bordák megvastagodása egy 3 éves betegnél, B csípő diszplázia egy másik 18 hónapos betegnél

MPS IH: karom kéz egy 18 hónapos betegben; B kézi röntgen.

MPS IH. a koponya kérgi csontjának megvastagodása és a sella turcica rendellenes “J-alakú” konformációja. A periventrikuláris terek tágulása b érzékben és c T-2 súlyozott mágneses rezonancia képek egy 17 hónapos betegről
típusú mukopoliszacharidózis (MPS II; OMIM # 309900) (ábra. 6), más néven Hunter-szindróma, az iduronát-2-szulfatáz (I2S) enzim hiányának eredménye, ami a DS és a HS GAG felhalmozódását eredményezi . A prevalencia körülbelül 1: 140 000-156 000 férfi élveszületés . Figyelemre méltó, hogy ez az egyetlen X-kapcsolt MPS, és a női hordozók általában tünetmentesek; azonban kivételesen érintettek lehetnek az X-kromoszóma rendellenességei, a homozigozitás vagy a ferde X-inaktiváció miatt . A fenotípusos expresszió a klinikai súlyosság széles spektrumát is lefedi. Súlyos esetekben jelentős progresszív neurológiai érintettség és szomatikus betegség együtt létezik, és a halál általában az élet második évtizedében következik be. Más, minimális vagy semmilyen neurológiai diszfunkcióval rendelkező betegek súlyos szomatikus érintettséget mutathatnak, míg a spektrum másik végén vannak olyan betegek, akiknek csak minimális szomatikus megnyilvánulása és normális intelligenciája van, akik túlélik a felnőttkort . A korai jeleket és tüneteket az MPS I és II súlyos formái osztják meg.

MPS II. 3 éves beteg, csak enyhe jellegzetes arcvonásokkal
a hydrops fetalis szórványos eseteit az MPS I írja le, bár általában ezek a gyermekek születéskor normálisnak tűnnek. Az MPS I első jelei általában az élet első hónapjaiban jelennek meg, míg az MPS II-ben lévők általában valamivel később jelentkeznek . Kiely et al. egy 55 Hurler betegen végzett retrospektív vizsgálatban megfigyelték, hogy a légzőszervi tünetek, a táplálkozási nehézségek, az inguinalis hernia és a középfülgyulladás 6 hónapos korban jelentkeztek, és a kyphosis, a szívbetegségek, a megnagyobbodott fej kerülete, a hypotonia, az organomegalia, a szaruhártya elhomályosodása, az ízületi korlátozás és a köldöksérv 6 és 12 hónap között jelentek meg. A köldöksérv az egyik legkorábbi bemutató funkció az MPS I-ben és II-ben, az inguinalis sérvekről pedig a betegek körülbelül 60% – ánál számoltak be .
az MPS I és II egy másik gyakori kezdeti nyom lehet a felső légúti visszatérő fertőzések fokozott váladékkal. A gyakori otitis, a krónikus visszatérő rhinitis és a tartós orrváladék nyilvánvaló fertőzés nélkül nem specifikusak, de segíthetnek a diagnosztikai gyanú megerősítésében, ha specifikusabb tünetekkel járnak. A GAG tárolása az oro-garatban a mandulák és az adenoidok megnagyobbodásához vezet, és hozzájárul a felső légúti szövődményekhez. Egy másik gyakori megállapítás a zajos légzés, különösen éjszaka, a későbbi szakaszokban obstruktív alvási apnoéval társul. A gyakori középfül-fertőzések és a középfül csontjainak dysostosisának következtében gyakori a halláskárosodás, mind vezetőképes, mind szenzorineurális hiányokkal . Gyakran megfigyelhető a Tracheobronchomalacia, amely akut légúti elzáródáshoz vagy összeomláshoz vezethet, ez a következő évek egyik fő halálokja .
az adenoidectomia, a tonsillectomia és a tympanostomia, valamint a sérv helyreállítása a gyakoribb és korai sebészeti beavatkozások az MPS I és II betegeknél, gyakran a diagnózis ismerete előtt (az esetek több mint 50% – ában) .
a visszatérő hasmenés meglehetősen gyakori az MPS I korai stádiumában, és neurológiai érintettségű MPS II betegeknél is jelen lehet, míg az enyhén érintett betegeknél nem gyakori .
Gibbus deformitás (dorso-ágyéki kyphosis) (ábra. 2) klinikailag nyilvánvalóvá válik 8 éves medián korban.6 hónaptól 1 évig az MPS I – ben, ami általában az MPS meglehetősen specifikus és korai jelzőjét képviseli. A csigolyatestek laposnak és csőrösnek tűnnek, ami későbbi szövődményekhez vezethet, mint például a gerincideg befogása, akut gerincsérülés és atlanto-occipitalis instabilitás; ezért különös figyelmet kell fordítani az atlanto-axiális ízület diszlokációjának megakadályozására általános érzéstelenítés és műtét során. 1 éves kor után az ízületi kontraktúrák és a genu valgum az ízületi károsodás és a progresszív csontváz diszplázia egyéb gyakori jeleit jelentik, amelyek 2 évnél korábban könnyen kimutathatók minden MPS által érintett csecsemőnél. Mindezeket a csontvázi megnyilvánulásokat, amelyek az MPS-re jellemzőek, dysostosis multiplexnek nevezik. A bordák megvastagodása röntgenfelvételeken látható (ábra. 3a), a hosszú csontok rövid, széles tengelyek, a térd hajlamos valgus, varus deformitások. Phalangealis dysostosis és szinoviális megvastagodása vezet jellegzetes karom kéz deformitás (ábra. 4), amely egy másik tipikus jellemző, amely felvetheti a betegség gyanúját. A csípő diszplázia gyakran az oka annak, hogy ezeket a gyermekeket először ortopéd szakemberek láthatják. Általában a medence rosszul alakul ki, a combfejek kicsiek, és a coxa valga gyakori, progresszív és gyengítő csípő deformációval (ábra. 3b).
jelentős különbség van az MPS I és az MPS II között; az MPS I-ben a csontváz növekedése 3 éves korig lassul, míg az MPS II betegek az első 5-6 éves korban túlnövekedést mutatnak , ezt követően lassítják növekedésüket .
az arcvonások (széles orr kiszélesedő orrlyukakkal, kiemelkedő szupraorbitális gerincekkel, nagy lekerekített arccal, vastag ajkakkal, megnagyobbodott kiálló nyelvvel) Durvulása, amelyet az orofacialis régió lágy szöveteiben és az arccsontokban történő Gag-Tárolás okoz , az első 2 évben nyilvánvalóvá válik, a Hurler-betegség medián életkorában 1,1 év, a Hunter-betegség súlyos formájában pedig 18 hónap és 4 év között, bár a finom fenotípusos változások korábban (már az első év második felében) észlelhetők speciális tapasztalattal rendelkező gyermekorvosok által (ábra. 1). A makrocefália fokozatosan nyilvánvalóbbá válik, a scaphocefaly pedig gyakori (ábra. 5), de a mikrocefália Hurler betegeknél is lehetséges . Az arc és a test hirsutizmusa mindig 24 hónapos korban nyilvánvaló . A bőr megvastagodhat és rugalmatlan lehet. Néhány MPS II betegnél jellegzetes bőrelváltozás alakul ki, amelyet elefántcsont-fehér papuláknak (kavicsszerűnek) neveznek a felkar felső részén és oldalán, a Hunter-szindróma patognomonikusaként .
a kardiomiopátia és a szelep szórólapok progresszív megvastagodása és merevedése mitrális és ritkábban aorta regurgitációhoz vezethet . A zörej az élet első hónapjaiban észlelhető, és néha a betegség első jeleként jelentik . A szív érintettsége szinte minden MPS II-ben szenvedő betegnél is jelen van, de ez körülbelül 5 éves korban kezdődik . A szelepbetegség jobb és bal kamrai hipertrófiához és szívelégtelenséghez vezethet.
a progresszív hepatosplenomegalia által okozott hasi kidudorodás 6 hónapos kor után könnyen nyilvánvaló, bár a gag-k tárolása a májban és a lépben nem vezet szervi diszfunkcióhoz .
a szaruhártya elhomályosodása szinte minden MPS I-ben szenvedő egyénnél 15 hónapnál korábban jelentkezik, ami súlyos látásromlást okozhat. Éppen ellenkezőleg, a szaruhártya elhomályosodása nem a Hunter-szindróma tipikus jellemzője, de a réslámpás vizsgálat diszkrét szaruhártya-elváltozásokat tárhat fel, amelyek nem befolyásolják a látást. Retina diszfunkció észlelhető, míg a glaukóma nem gyakori megállapítás.
a korai pszichomotoros fejlődést általában normálisnak észlelik, de az MPS I-ben a fejlődés késése általában 18 hónapos korra nyilvánvaló, a progresszív mentális hanyatlás súlyos értelmi fogyatékossághoz vezet. A nyelvi készségek nagyon korlátozottak ezekben a gyerekekben. Az MPS II fejlődési mérföldköveinek globális késése 2 év múlva nyilvánvalóvá válik . A súlyos Hunter-betegség vezető neurológiai jellegzetességeit azonban olyan markáns viselkedési zavarok jelentik, mint a hiperaktivitás, a makacsság és az agresszivitás, amelyek jellemzően nem figyelhetők meg az attenuált fenotípusúaknál .
a VII-es típusú Mucopolysaccharidosis (MPS VII; OMIM # 253220), más néven Sly-szindróma, egy rendkívül ritka MPS, amelyet a .. -glükuronidáz hiányos aktivitása jellemez, a kondroitin-szulfát (CS), DS és HS lizoszomális tárolásával, ami sejt-és szervi diszfunkcióhoz vezet. Az első beteget Sly et al. 1973 – ban eddig összesen 145 betegről számoltak be a szakirodalomban.
az MPS VII klinikai megjelenése és a betegség progressziója széles súlyossági spektrumot ölel fel, a korai, súlyos, multisystem megnyilvánulásoktól és az első hónapokban bekövetkezett haláltól kezdve egy enyhébb fenotípusig, amely később jelentkezik, normális vagy majdnem normális intelligenciával és hosszabb túléléssel jár .
az MPS VII-ben szenvedő betegek fenotípus-jellemzői hasonlítanak az MPS I és MPS II-hez (alacsony termet, csontváz dysplasia, macrocephaly, gingivális hypertrophia, visszatérő fülfertőzések, hepatosplenomegalia, hernias, cardialis érintettség, csökkent tüdőfunkció és kognitív károsodás). A leírt betegek váratlanul magas aránya (41%) kórtörténetében nem immun újszülött hydrops fetalis (NIHF) szerepel . Annak ellenére, hogy ezeknél a betegeknél a betegség prenatális kezdete volt, 13-ból 23 túlélte a csecsemőkort enyhe vagy közepes lefolyással késő tizenévesekké. Így a NIHF jelenléte önmagában nem jósolja meg a klinikai lefolyás lehetséges súlyosságát, ha a beteg túléli a csecsemőkort. Egy másik korai jel a macrocrania, míg a szívbillentyű érintettsége és a felső és alsó légúti ismételt fertőzései az élet első 2 évében jelentkeznek. A mérsékelt mentális retardáció és a beszédkárosodással járó progresszív halláskárosodás szintén nyilvánvaló az idő múlásával.
összefoglalva, a súlyos formái MPS van egy nagyon korai multisystem kezdet (6 hónapon belül az élet); Az MPS II hasonló, de később jelentkezik, és az MPS VII prenatális kezdete gyakori NIHF-vel és a felső és alsó légutak korai súlyos érintettségével jár.
többnyire neurológiai és kognitív érintettséggel rendelkező MPS
III típusú Mukopoliszacharidózisok (MPS III, más néven Sanfilippo szindróma; ábra. 7) van egy elterjedt neurológiai bemutatása. Az MPS III egy lizoszomális tárolási rendellenesség, amelyet a HS katabolizmusában részt vevő négy enzim egyikének hiánya okoz. Négy különböző altípusa ismert—MPS III típusú a (OMIM #252900), B típusú (OMIM #252920), C típusú (OMIM #252930), és D típusú (OMIM #252940)—mindegyik miatt különböző enzimhiány: a típusú, szulfamidáz/SGSH gén; B típusú, alfa-n-acetilglükozaminidáz/NAGLU gén; C típusú, alfa-glükozaminid N-acetiltranszferáz/HGSNAT gén; D típus, n-acetilglükozamina-6-szolfato sulfatasi/GNS gén). Mind a négy típus (A-tól D-ig) autoszomális recesszív öröklődéssel rendelkezik. Az MPS III a leggyakoribb az MPS közül, becsült prevalenciája 0,3 és 4 között van.1 eset minden 100 000 újszülöttre, az altípustól és a fajtól függően .

MPS III. Ugyanaz a beteg 2,5 éves és 5 éves korában. Vegye figyelembe a különböző arckifejezéseket, amelyek a neurológiai károsodás progresszióját mutatják
a Sanfilippo-szindrómában szenvedő betegek progresszív kognitív károsodást mutatnak az élet második és harmadik évében. Az MPS többségével ellentétben a szomatikus jellemzők viszonylag kevésbé nyilvánvalóak, a központi idegrendszer érintettsége pedig szembetűnő. A beszédfejlődés késése általában az első jel, amelyet a szülők észlelnek, de a fő aggodalmak lehetnek viselkedési problémák (hiperaktivitás, szorongó és agresszív viselkedés, alvászavarok), amelyet progresszív mentális hanyatlás követ . Ezek a tünetek téves diagnózist okozhatnak, mint viselkedési zavarok, figyelemhiányos hiperaktivitási zavar (ADHD) és autizmus spektrum zavarok . Epilepszia is jelen lehet.
az alvászavarok, amelyek előfordulási gyakorisága 80-90% – ig terjed , közös jellemző. Ezek az elalvás és a gyakori éjszakai ébredés nehézségeiből állnak, egyes betegeknél a nappali–éjszakai ritmus teljes megfordításával.
ezeket a megnyilvánulásokat nagyon nehéz kezelni, és óriási pszichológiai hatással vannak az egész család életminőségére.
általában, bár a fenotípus nagyon hasonló lehet a négy altípusban, a Sanfilippo B és C klinikai lefolyását kevésbé súlyos fenotípus jellemzi .
a nem neurológiai tünetek általában kevésbé kifejezettek az MPS III-ban, mint a többi MPS-ben. A visszatérő fül -, orr-és torokfertőzések fiatalabb korban figyelhetők meg. A visszatérő otitis, a középfül csonthibái és a belső fül rendellenességei süketséget okozhatnak. A túlzott vastag váladék és anatómiai változások a légutak elzáródását okozhatják. Az élet első éveiben gyakran jelentettek hasmenést, míg a székrekedés gyakoribb az idősebb betegeknél.
a fizikai vizsgálat durva arcvonásokat mutathat, bár ezek kevésbé hangsúlyosak. A betegek macrocephaly, széles szemöldök mediális fellángoló és synophrys, a haj általában száraz és durva, és hypertrichosis gyakori. Fiatalabb betegeknél enyhe hepatomegalia, valamint köldökzsinór és inguinalis herniák gyakran megfigyelhetők. Ritka, gyakran enyhe kontraktúrák többnyire a könyökben találhatók. A növekedést a 12 évesnél idősebb betegek körülbelül fele befolyásolja .
Összefoglalva, az MPS III vagy a Sanfilippo-betegség kiemelkedő neurológiai érintettséggel rendelkezik, nagyon enyhe szomatikus tünetekkel. A 2-3 éves kisgyermekek, akiknél hiperaktivitás, ADHD és agresszív viselkedés alakul ki, gyanúja lehet az MPS III-nak.
MPS elterjedt szomatikus érintettséggel
az MPS IV és az MPS VI csak szomatikus érintettséggel rendelkezik. Ezek olyan progresszív állapotok, amelyek megkímélik az intellektuális képességeket, és elsősorban a csontvázat (MPS IVA) vagy a test összes szervét/rendszerét (MPS VI) érintik. Az öröklés autoszomális recesszív. A tünetek súlyosbodásának sebessége az érintett egyének között változik.
Mucopolysaccharidosis IV (MPS IV, más néven Morquio szindróma; ábra. 8) két genetikailag különböző rendellenességként jelenik meg, amelyek mindegyike eltérő enzimhiánnyal jár: MPS IVA (Morquio a szindróma; OMIM #253000) az N-acetil-galaktozamin-6-szulfatáz (GALNS gén) hiánya miatt, ami keratan-szulfát (KS) és CS felhalmozódását eredményezi ; és MPS IVB (Morquio B szindróma; OMIM #253010) a béta-galaktozidáz aktivitás hiánya miatt, ami a vizelet KS-hez és oligoszacharid kiválasztódáshoz vezet, mint a GM1 betegeknél. Az MPS IVB valóban allélikus a GM1-gangliosidózis különféle formáira, de hiányzik a GM1 gangliosidózisban tapasztalt pszichomotoros romlás .

MPS IVA. egy 4 éves beteg b antero-posterior és C laterális röntgensugarakkal, amelyek “lapát alakú” bordákat és lapított és lekerekített csigolyákat mutatnak, tipikus “elülső csőr” aspektussal
a súlyos MPS IVA-ban szenvedő betegek, akiknek nincs kimutatható enzimaktivitása, első tüneteiket általában az első életévben mutatják be, bár ritkán diagnosztizálják őket 4-5 éves kor előtt. A Morquio a betegek természetes kórtörténetére vonatkozó információk főként két széles körű adatgyűjtésből származnak . A csökkent növekedési ütem körülbelül 18 hónapos korban kezdődik, és a növekedés körülbelül 7 vagy 8 éves korban megáll. A többi MPS-szel ellentétben a Morquio a ízületek általában lazák és nagyon rugalmasak (hipermobil), de egyes ízületek (általában a csípő és a váll) korlátozott mozgástartományúak lehetnek. Az ilyen csontrendszeri érintettséggel rendelkező betegek diagnosztizálhatják vagy értékelhetik spondyloepiphysealis dysplasia, pseudoachondroplasia, többszörös epiphysealis dysplasia vagy bilaterális Legg-Calv Kb-Perthes betegség .
állandó jellemző az odontoid hypoplasia; így az atlanto-occipitalis ízület diszlokációja előfordulhat, ami kompressziót és a bulbar és a gerincvelő károsodását okozhatja, ami bénulást vagy akár halált is okozhat, ha a nyaki stabilizációt nem hajtják végre Korán.
az MPS IVB betegek nagyon hasonló klinikai megnyilvánulással rendelkeznek, de kevésbé kiemelkedő alacsony termetűek. Enyhén durva arcvonásokkal, halláskárosodással, szaruhártya-homályossággal, szívbillentyű-betegséggel és gyakori felső légúti fertőzésekkel rendelkeznek. Ők is jelen lágyéksérv és enyhe hepatomegalia. A csontváz diszplasztikus jellemzői közé tartozik a platyspondyly, az odontoid hypoplasia lehetséges nyaki subluxációval, kypho-scoliosis, coxa valga, valamint a szűkített csípő szárnyak .
Mucopolysaccharidosis VI (MPS VI, más néven Maroteaux-Lamy szindróma; OMIM # 253200; ábra. 9) az 5.kromoszómán található arilszulfatáz B (ARSB) gén patogén mutációinak köszönhető. Ennek következtében az arilszulfatáz B (ASB) enzim (N-acetil-galaktózamin-4-szulfatáz) csökkent vagy hiányzó aktivitása rontja a DS lebomlását, meghatározva annak sejtfelhalmozódását .

MPS VI. egy 2 éves fiú nyilvánvaló durva arccal és csontváz dysostosissal
azoknál a betegeknél, akiknek nincs kimutatható enzimaktivitása, az MPS VI súlyos formája van, és korai gyermekkorban tüneteket mutatnak. Szomatikus fenotípusuk hasonló a Hurler-szindrómához. A legtöbb esetben 2 vagy 3 éves korig nyilvánvalóvá válik a dysostosis multiplexnek nevezett súlyos és progresszív csontvázkárosodás. Ez a csontrendszeri diszplázia magában foglalja a combcsont diszpláziás diszpláziáját, a rendellenes csigolyatesteket, a szabálytalan kulcscsontokat, a hipoplasztikus disztális singcsontot és sugarat, valamint a diszplasztikus és rövid metakarpális csontokat; a carpalis és tarsalis csontok hipoplasztikusak és szabálytalan profilúak. A növekedési sebesség az első életév után gyakran lelassul, a végső magasság általában 120 cm-nél alacsonyabb. Mint más képviselőknél, a korai jel a durva arcvonások. Ezen túlmenően, a betegek egy tipikus kiálló has, hepatomegalia, köldökzsinór és / vagy lágyéksérv, és a szív bevonása korai csecsemőkorban. Bár elsődleges értelmi fogyatékosság nincs jelen, néhány betegnél hydrocephalus figyelhető meg, ennek következtében intracranialis hypertensio és papilledema alakul ki. A Pectus carinatum, a merev és összehúzódott ízületek, a scoliosis és a kyphosis, valamint a nyaki gerincvelő kompressziója idővel romlik .
Összefoglalva, az MPS IV és VI nem mutat központi idegrendszeri érintettséget. Az MPS IV egyedülálló MPS az ízületi lazaság bemutatásában, míg a súlyos MPS VI megjelenése hasonló az MPS I.