Autosomica Recessiva Congenite Ittiosi | Actas Dermo-Sifiliográficas
Introduzione
L’ultima consenso classificazione di ittiosi distingue tra 2 forme principali: le forme sindromiche, che presentano con manifestazioni cutanee non solo, e le forme sindromiche, che presentano con manifestazioni in altri organi (Tabella 1).1 Tra le forme non sindromiche, vengono identificati 4 gruppi: ittiosi comuni, ittiosi congenita autosomica recessiva (ARCIs), ittiosi cheratinopatiche e altre ittiosi meno comuni.Tradizionalmente, il gruppo di ARCIs era diviso in 2 disturbi, ittiosi lamellare (LI) e eritroderma ittiosiforme congenito (CIE). Nella nuova classificazione, l’ittiosi arlecchino (HI) è stata aggiunta a questo gruppo1 perché mutazioni inattivanti nel gene ABCA12 sono state identificate come responsabili di questo disturbo,2,3 mentre mutazioni nonsense nello stesso gene possono dare origine al fenotipo LI4 o CIE5,6. Altre varianti meno comuni incluse nel gruppo di ARCIs sono l’auto-guarigione collodion baby (SHCB), l’acrale SHCB e l’ittiosi da bagno.7-9
Classificazione di consenso basata sulle caratteristiche cliniche dell’ittiose1.
| Nonddromic Forme | Forme Sindromiche |
| Comune IchthyosesIchthyosis vulgarisRecessive x-linked ittiosi (non sindromica )ajimajor formsHarlequin ichthyosisLamellar ichthyosisCongenital ichthyosiform erythrodermaMinor formsSelf-guarigione collodio babyAcral di auto-guarigione collodio babyBathing soddisfare ichthyosisKeratinopathic IchthyosesMajor formsEpidermolytic Ichthyosissuperficial epidermolytic ichthyosisminor formsannular epidermolytic ichthyosiscurth-Macklin ichthyosisautosomal recessivo epidermolytic ichthyosisEpidermolytic nevusOther FormsLoricrin keratodermaErythrokeratodermia vararabilispeeling pelle syndromeCongenital reticolare ichthyosiform erythrodermaKLICK sindrome | Sindromiche X-linked Ichthyosrecessive x-linked ittiosi (sindromiche)Ittiosi follicularis, alopecia, e fotofobia (IFAP) syndromeconradi-Scammann-happle sindrome (chondrodysplasia punctata di tipo 2)sindromica autosomica ichthyosisskin disordersnetherton syndromeichthyosis-hypothrichosis syndromeichthyosis-Colangite Sclerosante syndrometrichothystrophyneurological Disorderssjögren-Larsson syndromeRefsum diseaseMEDNIK syndromeFatal disease courseGaucher disease, type 2Multiple sulfatase deficiencyCEDNIK syndromeARC syndromeOther associated signsKID syndromeChanarin-Dorfman syndromeIchthyosis prematurity syndrome |
Abbreviations: ARC, arthrogryposis–renal dysfunction–cholestasis; ARCI, autosomal recessive congenital ichthyosis; CEDNIK, cerebral dysgenesis, neuropathy, ichthyosis, and palmoplantar keratoderma; KID, keratitis ichthyosis deafness; KLICK, keratosis linearis with ichthyosis congenital and sclerosing keratoderma; MEDNIK, ritardo mentale, enteropatia, sordità, neuropatia periferica, ittiosi, cheratoderma.
Sono disponibili solo dati limitati sull’epidemiologia di ARCIs. Negli Stati Uniti, è stata stimata una prevalenza alla nascita di 1 per 100 000 abitanti per LI e di 1 per 200 000 abitanti per CIE. Altri studi hanno riportato una prevalenza combinata per LI e CIE di 1 per 200 000 a 300 000 popolazione.10,11 In alcuni paesi come la Norvegia, la prevalenza stimata è maggiore (1 per 91 000) a causa delle mutazioni del fondatore.12 Il ritrovamento di 1 o più mutazioni ricorrenti in una popolazione può essere dovuto al fatto che la mutazione si è verificata in un dato punto della storia ed è stata quindi trasmessa di generazione in generazione (mutazione del fondatore) o perché la regione del genoma in cui si trova la mutazione ha una sequenza di DNA suscettibile alla mutazione (mutazione hotspot). In Spagna, la prevalenza stimata di ARCI è 1 ogni 138 000 nella popolazione generale e 1 ogni 61 700 tra i bambini sotto i 10 anni di età.13 In alcune regioni della Spagna, la prevalenza potrebbe essere ancora più elevata. Sulla costa galiziana, ad esempio, è stata segnalata una prevalenza di 1 per 33 000, dovuta anche a un effetto fondatore.14
Ittiosi lamellare e Caratteristiche eritrodermacliniche ittiosiformi congenite
Sebbene originariamente si pensasse che LI e CIE fossero entità diverse, ci sono state segnalazioni di pazienti con manifestazioni cliniche intermedie ed entrambe le condizioni possono essere causate da mutazioni nello stesso gene.15,16 Inoltre, i pazienti con la stessa mutazione, anche all’interno della stessa famiglia, possono sviluppare fenotipi diversi.12,15
La maggior parte dei pazienti nasce avvolta da una membrana di collodio che scompare progressivamente durante le prime settimane di vita e viene sostituita dal fenotipo definitivo (Fig. 1 BIS). Ipoidrosi, grave intolleranza al calore e distrofia delle unghie sono frequentemente osservate sia in LI che in CIE.17-19 Pazienti con LI di solito hanno manifestazioni cliniche più gravi rispetto a quelli con CIE. Hanno grandi squame platelike, spesso di un colore scuro, che copre l’intera superficie del corpo. L’eritroderma è assente o minimo. Tali pazienti di solito hanno ectropion e, a volte, eclabium, ipoplasia della cartilagine articolare e nasale, alopecia cicatriziale, specialmente sul bordo del cuoio capelluto, e cheratoderma palmoplantare (Fig. 1B e C). La CIE è caratterizzata dalla presenza di eritroderma e scaglie biancastre fini (Fig. 2). Alcuni pazienti hanno marcato eritema e ridimensionamento generalizzato. Le squame possono essere grandi e di colore scuro, in particolare sulle superfici estensori delle gambe. Nei casi meno gravi, l’eritema è lieve e il ridimensionamento va bene.

Caratteristiche cliniche dell’ittiosi lamellare. A, desquamazione lamellare brunastra. B, ipercheratosi plantare marcata. C, alopecia cicatriziale del cuoio capelluto.
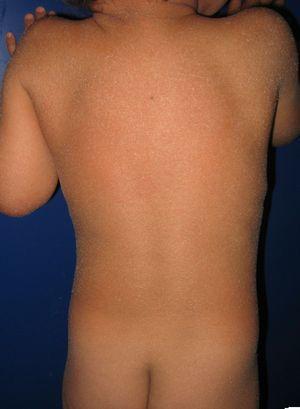
Paziente con eritroderma ittiosiforme congenito e mutazioni nel gene ALOXE3. Si può osservare un lieve eritema e una desquamazione furfuracea biancastra generalizzata.
Istopatologia
I cambiamenti istopatologici non forniscono una diagnosi. In LI si osserva una massiccia ipercheratosi ortocheratotica, di solito con il doppio dell’estensione come in CIE. L’epidermide è acantotica e occasionalmente assume un aspetto simile alla psoriasi. Il tasso di proliferazione cellulare è normale o leggermente elevato.17-19 Pazienti con CIE hanno ipercheratosi meno marcata, con paracheratosi focale o estesa, uno strato granulare normale o ispessito e acantosi più pronunciata. Il turnover epidermico è aumentato.17-19
Ultrastruttura
Sebbene non sia stata finora trovata una stretta correlazione tra risultati molecolari, clinici e ultrastrutturali, la microscopia elettronica può tuttavia essere utile per escludere altre forme di ittiosi e per guidare le analisi genetiche in alcuni casi. Sono stati descritti quattro tipi di ittiosi congenita (Tabella 2).
Classificazione ultrastrutturale delle ittiosi congenite.
| Tipo | Caratteristica Principale | Altre Caratteristiche | Mutazioni | Manifestazioni Cliniche |
| 1 | l’Assenza di ultrastrutturali marcatori di ittiosi tipi 2, 3, e 4 | goccioline di Lipidi o anelli nello strato corneo (il più frequente), Piccolo keratohyalin granulesVesicular o lobulare membrana di rivestimento granuli | TGM1 (33.3%)ALOX12B (2 casi) | CIE |
| 2 | crepacci di Colesterolo nello strato corneo | Assenza o diradamento dei cornified envelopeSmall keratohyalin granulesLipid goccioline | TGM1 (89-100%) | LI |
| 3 | Laminato membraneous strutture nello strato granuloso e/o strato corneo. | Anormale membrana di rivestimento granulesLipid dropletsFoci di spicco juxtanuclear vacuoli strato granulare | NIPAL4 (93%) | CIE (più frequente), LI |
| 4 | Trilamellar membrana pacchetti di colmare alcune cellule nello strato granuloso e/o strato corneo | Anormale membrana di rivestimento granuli | FTAP4 | Ittiosi prematurità, sindrome di (100%) |
Abbreviazioni: CIE, congenita ichthyosiform erythroderma; LI, ittiosi lamellare.
Ittiosi congenita di tipo 1
L’ittiosi congenita di tipo 1 è caratterizzata dall’assenza di marcatori ultrastrutturali per i tipi di ittiosi 2, 3 e 4. Pertanto, la diagnosi viene solitamente effettuata solo quando gli altri tipi sono stati esclusi. La scoperta più frequente è la presenza di goccioline lipidiche o anelli nello strato corneo (Fig. 3 BIS).20 Queste goccioline lipidiche non sono una caratteristica costante o specifica di questo particolare tipo in quanto non sono presenti in tutti i casi,20 e possono essere presenti in altri tipi di ittiosi.21,22 Clinicamente, la maggior parte dei pazienti presenta manifestazioni di CIE.12,20 Un terzo dei pazienti ha mutazioni nel gene TGM1.16 Questo tipo ultrastrutturale è stato anche identificato in associazione con mutazioni nel gene ALOX12B.23,24

Immagini al microscopio elettronico. A, ittiosi congenita di tipo 1, che mostra goccioline lipidiche nello strato corneo e assenza di marcatori ultrastrutturali degli altri tipi di ittiosi. B, ittiosi congenita di tipo 2, caratterizzata dalla presenza di fessure di colesterolo (freccia) nei corneociti.
Ittiosi congenita di tipo 2
L’ittiosi congenita di tipo 2 è caratterizzata da fessure di colesterolo nello strato corneo (Fig. 3 TER).21 Tali fessure sono una constatazione costante in questo tipo di ittiosi e possono essere rilevate in diverse biopsie nello stesso paziente; il trattamento con retinoidi orali non ha alcun impatto su queste fessure.12,25 Aggregati elettron-densi sono stati osservati anche sui corneociti in alcuni pazienti con attività TGase 1 carente.26-28 Clinicamente, la maggior parte dei pazienti presenta gravi manifestazioni di CIE.12 Questo tipo ultrastrutturale è fortemente associato a mutazioni nel gene TGM1.12,16
Ittiosi congenita di tipo 3
L’ittiosi congenita di tipo 3 è caratterizzata da strutture membranose lamellari nello strato granuloso e/o nello strato corneo. Queste strutture sono disposte in strisce attorno a uno spazio vuoto vicino al nucleo.22,29-31 Le manifestazioni cliniche in questo tipo sono diverse dalle altre; l’insorgenza dell’ittiosi è variabile, la desquamazione e l’eritema possono essere irregolari o generalizzati e le flessioni in particolare sono influenzate. Le mutazioni nel gene NIPAL4 sono responsabili del 93% delle ittiosi di tipo 3.32
Ittiosi congenita Tipo 4
Tipicamente, nell’ittiosi congenita tipo 4, alcune cellule nello strato granuloso e nello strato corneo sono riempite con pacchetti di membrana trilamellare.33 Questi risultati sono patognomici per la sindrome da prematurità di ittiosi, una condizione attualmente considerata come una forma sindromica di ittiosi.34,35
Studi molecolari
In termini genetici, gli ARCIS sono molto eterogenei. Il gene TGM1 è associato alla maggior parte dei casi, ma sono state riportate mutazioni in altri 5 geni (ALOX12B, ALOXE3, NIPAL4, CYP4F22 e ABCA12). Fischer et al.36 ha studiato 520 famiglie con ARCI e ha identificato mutazioni in almeno 1 di questi geni nel 78% dei casi (TGM1 nel 32%, NIPAL4 nel 16%, ALOX12B nel 12%, CYP4F22 nell ‘ 8%, ALOXE3 nel 5% e ABCA12 nel 5%). In un altro studio su 250 pazienti con ARCI di diversa origine, il 38% aveva mutazioni TGM1, il 6,8% aveva mutazioni ALOXE3 e il 6,8% aveva mutazioni ALOX12B.37 In Galizia, abbiamo identificato mutazioni nei geni TGM1, ALOX12B, ALOXE3, NIPAL4 e CYP4F22 nel 75% delle famiglie studiate, ma la distribuzione delle mutazioni era diversa.14 Il gene TGM1 è stato mutato in 68.7% dei casi mentre il gene ALOXE3 è stato mutato in solo 1 paziente. Non abbiamo rilevato mutazioni in nessuno degli altri 3 geni studiati.
TGM1
Il gene TGM1 si trova sul cromosoma 14q11.2 e ha 15 esoni (GenBank NM-000359.2). Codifica l’enzima TGase 1, che è uno dei 3 enzimi TGase trovati nell’epidermide.38 Questo enzima partecipa alla formazione dell’involucro cornificato catalizzando il cross-linking calcio-dipendente di diverse proteine come involucrina, loricrina e proteine ricche di prolina.39,40 Catalizza anche il legame di ??- idrossiceramidi nello strato esterno dell’involucro cornificato con proteine nello strato interno.41,42 Nei pazienti con mutazioni TGM1, l’involucro cornificato è mancante e l’attività della TGase 1 è ridotta o inesistente.43-47
Dal 1995, quando questo gene è stato identificato come responsabile di alcuni casi di ARCI,48-50 più di 110 mutazioni sono state riportate in pazienti di diversa origine. Le mutazioni in TGM1 sono la causa più comune di ARCI.36,37 Questa mutazione è stata trovata nel 55% dei casi negli Stati Uniti e nell ‘ 84% dei casi in Norvegia.12,51 La mutazione più frequente è c.877-2A > G, che è stato trovato nel 34% degli alleli mutati riportati fino ad oggi.52 L’alta frequenza di questa mutazione in paesi come gli Stati Uniti e la Norvegia è dovuta ad un effetto fondatore.12,53 La seconda mutazione più frequente è p.Arg142His. Questa e simili mutazioni sono state riportate in paesi come Egitto, Germania, Finlandia e Stati Uniti, 15,49-51,54-56 e sembrerebbe che queste siano mutazioni hotspot.57 La mutazione p. Arg307Trp è frequente nella popolazione giapponese.5 In Galizia, il p.Arg760X, c. 1223_1227delACACA e c.984 + 1G>Mutazioni A in TGM1 sono state identificate nell ‘ 81,82% delle famiglie con mutazioni in questo gene, suggerendo un effetto fondatore.14 La conferma di questa ipotesi è stata ottenuta dallo studio dell’aplotipo (lavoro ancora inedito).
Le mutazioni TGM1 sono responsabili della maggior parte dei casi di LI15,27,44,46,56,58-63 e per una piccola percentuale di casi di CIE.43,47,64,65 Tali mutazioni possono anche dare origine ad altre forme di ARCI come SHCB, SHCB acrale e ittiosi da bagno.
Molti studi hanno tentato di dimostrare associazioni genotipo-fenotipo tra mutazioni in TGM1 e risultati ultrastrutturali o clinici, ma fino ad oggi non è stata osservata alcuna correlazione significativa.15,16,53 In generale, i pazienti con mutazioni nel gene TGM1 sono più gravemente colpiti rispetto a quelli senza tali mutazioni. In uno studio su 83 pazienti con ARCI in Svezia ed Estonia, la presenza di ectropion e collodion baby è stata associata a mutazioni TGM1, mentre un più alto tasso di eritema è stato osservato in pazienti senza mutazioni in questo gene.66 Un altro studio ha dimostrato che il tipo di ridimensionamento è la principale differenza tra portatori e non portatori di mutazioni TGM1, scoprendo che tutti i pazienti con mutazioni in questo gene avevano una scalatura lamellare mentre l ‘ 80% di quelli senza mutazioni TGM1 aveva una scalatura fine.14 Inoltre, è stato visto che le mutazioni troncanti sono più frequentemente associate a ipoidrosi e disturbi della sudorazione rispetto alle mutazioni missense.51 Nella popolazione nordamericana, un modello basato sulla presenza di alcune caratteristiche cliniche predice che i pazienti che sono nati come bambini di collodio e hanno disturbi oculari e/o alopecia hanno 4 volte più probabilità di avere mutazioni TGM1.51
ALOXE3 e ALOX12B
I geni ALOXE3 e ALOX12B si trovano sul cromosoma 17p13.1.67 Hanno una struttura simile con 15 esoni che codificano i LOX epidermici eLOX-3 e 12R-LOX.68,69 Il fatto che siano prevalentemente espressi negli strati soprabasali dell’epidermide supporta il loro ruolo nelle fasi avanzate di differenziazione epidermica, con partecipazione alla lavorazione dei corpi lamellari.24,70 Questi enzimi agiscono su passi adiacenti nella via dell’epoxilina (Fig. 4). 12R-LOX trasforma l’acido arachidonico in acido 12R-idrossieicosatetraenoico mentre eLOX-3 converte questo prodotto in un isomer69,71 epossialcohol della famiglia hepoxilin A3.72 Il prodotto di hepoxilin è instabile ed è idrolizzato in celle ad un derivato triidrossi specifico (trioxilin). Sebbene il ruolo esatto dei prodotti della via dell’epoxilina non sia noto, è stato ipotizzato che possano partecipare alla formazione di lipidi intercellulari dello strato corneo o agire come segnali per indurre la differenziazione dei cheratinociti.
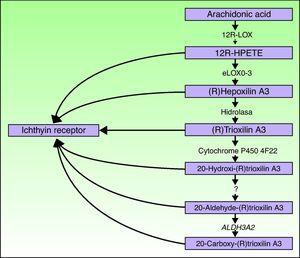
Diagramma schematico della via epoxilina, che mostra la partecipazione dei geni ALOXE3, ALOX12B, NIPAL4 e CYP4F22. Le mutazioni in questi geni sono responsabili di alcuni tipi di ARCI. HPETE indica acido idroperossieicosatetraenoico.
I geni ALOX12B e ALOXE3 sono stati identificati per la prima volta nel 2002.73,74 Da allora, sono state riportate più di 30 mutazioni nei geni ALOX12B23,24,37,75-77 e circa 10 nei geni ALOXE337,74,75. Queste mutazioni sono responsabili del 14-17% di ARCIs36,37 e 72.2% di SHCB.23,78,79 La relazione causale tra queste mutazioni e fenotipo è stata confermata dimostrando che l’attività catalitica del LOX epidermico è stata totalmente abolita nei pazienti con queste mutazioni75,80 e utilizzando modelli animali che riproducevano il fenotipo ittiosiforme visto nell’uomo.81-83 Entrambi i geni sono responsabili di una percentuale simile di casi ARCI. Tuttavia, la gamma di diverse mutazioni nel gene ALOXE3 è limitata, a causa della predominanza di 2 mutazioni, p. Arg234X e p. Pro630Leu, che sembrano corrispondere agli hotspot.37,74,75
I pazienti con mutazioni nei geni ALOXE3 e ALOX12B di solito mostrano un fenotipo CIE.74,75,77 La gravità del ridimensionamento è lieve o moderata e le squame hanno un colore biancastro o marrone chiaro. Può anche essere presente eritema. Ben il 76% dei pazienti nasce come collodio e l ‘ 88% ha disturbi della sudorazione.37 Pazienti con mutazioni nel gene ALOX12B mostrano una desquamazione biancastra più limitata rispetto ai portatori di mutazioni nel gene ALOXE3. In questi casi, le squame sono brunastre e aderenti. La presenza di eritema, ipercheratosi palmoplantare e accentuazione delle pieghe palmoplantari sono anche associate a mutazioni di ALOX12B.37
Ichthyin/NIPAL4
Il gene NIPAL4, noto anche come gene ichthyin, si trova sul cromosoma 5q33. Ha 6 esoni che codificano una proteina con diversi domini transmembrana di funzione sconosciuta.84 È stato ipotizzato che il prodotto proteico partecipi alla stessa via metabolica del LOX e possa agire come recettore per le trioxiline A3 e B3 o per altri metaboliti della via metabolica dell’epoxilina.84 Sarebbe quindi implicato nella formazione di corpi lamellari o nel loro trasporto verso lo spazio extracellulare.32 A sostegno di questo sono 2 osservazioni. Innanzitutto, nel 93% dei casi, le mutazioni in questo gene sono associate a un pattern ultrastrutturale di ittiosi congenita di tipo 3, caratterizzato da anomalie nei corpi lamellari e dalla presenza di membrane perinucleari allungate nello strato granuloso.32 Secondo, NIPAL4 è espresso essenzialmente nello strato granuloso dell’epidermide, dove sono presenti i corpi lamellari.85
Dalla scoperta del gene NIPAL4 nel 2004,84 solo 9 mutazioni sono state riportate in pazienti provenienti da paesi mediterranei (Algeria, Turchia e Siria), 84 paesi scandinavi,32 Pakistan,85 Isole Faroe,32 e Sud America.84
Lo spettro clinico dei pazienti con mutazioni in questo gene è ampio, anche tra i membri della stessa famiglia. Tra il 3,7% 32 e il 60% 84 nascono come collodio. Quando la membrana di collodio scompare, la maggior parte dei pazienti sviluppa le manifestazioni di CIE, con scaglie biancastre fini su una base eritematosa sul viso e sul tronco e scaglie brunastre più grandi sul collo, sulle natiche e sulle gambe.Xerosi marcata, placche ipercheratotiche reticolari brunastre generalizzate che appaiono accentuate nelle pieghe della pelle e possono essere presenti discromie facciali.32,85 Inoltre, il cheratoderma palmoplantare è una scoperta frequente insieme a contratture occasionali delle dita e unghie ricurve. Alcuni studi hanno riportato risultati più tipici di LI.32,85 La presenza di segni e sintomi di dermatite atopica è stata riportata in alcuni pazienti, sebbene mutazioni nel gene FLG non siano state rilevate in nessuno di questi casi.85
CYP4F22
Il gene FLJ39501 o CYP4F22 si trova sul cromosoma 19p13.12.86 Ha 12 esoni 87 e codifica un citocromo P450, famiglia 4, sottofamiglia F, polipeptide 2, omologo di leucotriene B4 – ω-idrossilasi (CYP4F2). La reazione catalizzata dal prodotto di FLJ39501 nella pelle e nei substrati di tale reazione può essere dedotta per analogia con i suoi omologhi noti CYP4F2 e CYP4F3.88 È stato ipotizzato che CYP4F2 e CYP4F3 partecipino alla via dell’epoxilina catalizzando la conversione della trioxilina A3 in 20-idrossi-(R)trioxilina A387 e che il prodotto finale di questa via, 20-carbossi-trioxilina A3, possa avere un effetto regolatore biologico chiave nella pelle.89
Ad oggi, solo 8 mutazioni di questo gene sono state segnalate in 12 famiglie consanguinee dei paesi mediterranei87 e in 1 famiglia di origine israeliana.62
Nelle famiglie riportate da Lefèvre et al., 87 la maggior parte pazienti ha avuti un fenotipo di CIE alla nascita e questo successivamente è progredito a LI. I pazienti sono stati sopportati solitamente con eritroderma marcato, sebbene senza alcuna membrana di collodio. Man mano che invecchiavano, svilupparono una desquamazione grigio-biancastra generalizzata, che era più marcata nella regione periumbilicale, sui glutei e sulla parte inferiore del corpo. Iperlinearità dei palmi e delle piante dei piedi e desquamazione sul cuoio capelluto, a volte di tipo pitiriasiforme, erano frequenti.87 In un’altra famiglia, i 3 membri colpiti sono nati come collidion babies e hanno sviluppato eritroderma intenso, desquamazione generalizzata e cheratoderma palmoplantare.62
ABCA12
Nel 2003, il gene ABCA12 è stato segnalato per essere responsabile di alcuni casi di LI ed è stato mappato al cromosoma 2q34.4 È stato successivamente confermato che le mutazioni in questo gene erano anche responsabili di HI.2, 3ABCA12 codifica 53 esoni e appartiene ad una famiglia di trasportatori ABC, che legano l’adenosina trifosfato facilitando anche il trasporto di diverse molecole attraverso la membrana cellulare.90 I membri della sottofamiglia ABCA sono tutti implicati nel trasporto lipidico.91 La funzione carente di ABCA12 causa i disordini di trasporto del lipido nei corpi lamellari e così piombo ad una diminuzione nei livelli intercellulari del lipido nello strato corneo.3studi ultrastrutturali hanno dimostrato che ABCA12 si trova in corpi lamellari associati a glicosilceramidi.Le mutazioni di 91ABCA12 sono state associate a disturbi nella distribuzione e nel trasporto di glicosilceramidi e con livelli ridotti di idrossiceramidi, uno dei componenti principali della barriera lipidica negli spazi intercellulari.3,6,92,93 L’ipercheratosi massiccia che si verifica in questi pazienti potrebbe essere una risposta compensatoria a una barriera lipidica carente.94 Potrebbe anche essere dovuto alla mancanza di desquamazione dei corneociti,93 che potrebbe essere causata da difetti nel trasporto di alcune proteasi, come la callicreina 5 e la catepsina D, derivanti da disturbi nei corpi lamellari.95 Modelli murini e studi in vitro suggeriscono che le mutazioni ABCA12 hanno anche un effetto sulla differenziazione epidermica.95-97
Ad oggi, sono state riportate più di 50 mutazioni nel gene ABCA12 in pazienti con ARCI provenienti da Africa, Europa, Pakistan e Giappone. Le mutazioni più frequenti sono p. Val244SerfsTer28, 2, 98, 99 identificato in popolazioni pakistane e indiane,e p.Asn1380Ser, 4 identificato in famiglie africane. In entrambi i casi, queste possono essere mutazioni fondanti.
L’entità delle mutazioni ABCA12 è correlata al fenotipo, con mutazioni associate a completa perdita di funzione che portano al fenotipo HI.2,3,98–102 Al contrario, in LI e CIE, la maggior parte delle mutazioni sono missense e hanno un effetto meno grave sulla funzione proteica.4-6, 103 Le mutazioni alla base del fenotipo LI sembrano essere concentrate nella prima regione della cassetta di legame dell’adenosina trifosfato.4 Clinicamente, i pazienti con CIE e mutazioni nel gene ABCA12 hanno scale di medie dimensioni che sono leggermente più grandi di quelle normalmente osservate nei pazienti con questo fenotipo.
Arlecchino ittiosi
HI o arlecchino feto è una forma grave e di solito fatale di ittiosi. I bambini sono di solito prematuri con ampie placche ipercheratotiche lucide, separate da profonde fessure, che coprono l’intero tegumento e formano motivi geometrici che ricordano gli abiti indossati dagli arlecchini, dando così alla condizione il suo nome. La tenuta della pelle porta a una marcata eversione delle palpebre e delle labbra, allo sviluppo rudimentale della cartilagine articolare e nasale e, occasionalmente, alla microcefalia. I bambini raramente hanno ciglia o sopracciglia, anche se i capelli sul cuoio capelluto possono essere conservati. Le mani e i piedi sono gonfi ed edematosi e spesso coperti da uno strato simile a un guanto. Possono avere contratture delle dita.
Per tali pazienti, il rischio di morire durante il periodo neonatale è molto alto.104 La ventilazione polmonare è compromessa; la perdita di acqua transepidermica porta a disidratazione, squilibrio idroelettrico e instabilità termica; e il rischio di infezioni è aumentato. La tenuta facciale e l’eclabio ostacolano la suzione e quindi l’alimentazione, con il corrispondente peggioramento della disidratazione. I neonati con questa condizione raramente vivevano più a lungo alcune settimane. Negli ultimi anni, tuttavia, le possibilità di sopravvivenza a lungo termine sono aumentate notevolmente, essenzialmente a causa della somministrazione di retinoidi sistemici e dei progressi nelle cure neonatali intensive.In uno studio recente, l ‘ 83% dei pazienti trattati con retinoidi orali è sopravvissuto rispetto al 24% dei pazienti non trattati. La maggior parte dei decessi si è verificato nei primi 3 giorni di vita, ma il trattamento non è stato avviato fino a dopo questo in molti dei sopravvissuti.104 Ciò suggerisce che molte di queste morti precoci si sarebbero verificate indipendentemente dal trattamento con retinoidi.
I bambini che sopravvivono al periodo neonatale sviluppano generalmente CIE gravi.106 La natura e la posizione delle mutazioni nel gene ABCA12 e l’entità della perdita della funzione del trasportatore possono determinare la prognosi.3.92.107 Pazienti che conservano un certo grado di attività proteica, anche se minima, possono avere maggiori possibilità di sopravvivere. I portatori di mutazioni omozigoti hanno un tasso di mortalità più elevato.104
La principale caratteristica istologica dell’HI è la presenza di uno strato corneo ortocheratotico estremamente denso e compatto. I follicoli piliferi e i condotti del sudore hanno prominenti tappi ipercheratosici107, 108 e hanno corpi lamellari anormali o assenti, inclusioni lipidiche o resti di organelli o nuclei nei corneociti e assenza di lipidi intercellulari nello studio ultrastrutturale.108.109 I follicoli piliferi mostrano una marcata concentrazione di materiale cheratotico, che è una caratteristica diagnostica di HI utilizzata per la diagnosi prenatale.
Ad oggi, il tasso di rilevamento delle mutazioni nel gene ABCA12 nei pazienti con HI è vicino al 100%, e quindi questo sembrerebbe essere una condizione geneticamente omogenea.
Collodion Baby e Collodion Baby di auto-guarigione
I bambini Collodion di solito nascono prematuramente e la morbilità e la mortalità perinatale sono aumentate. Alla nascita, il neonato è coperto da una membrana trasparente insegnata lucida che ricorda l’involucro di cellophane (Fig. 5). I bambini hanno ectropion, eclabium e ipoplasia della cartilagine nasale e articolare. La suzione e la ventilazione polmonare possono essere ostacolate110 e la perdita transepidermica di acqua e il rischio di infezioni sono aumentati.110,111

Collodio bambino che successivamente progredito ad un fenotipo ittiosi lamellare.
Collodion baby è la solita presentazione per HI e CIE. Autosomica dominante LI, 112, 113 Sindrome di Sjögren-Larsson, 110 tricotiodistrofia, 114 malattia giovanile di Gaucher, 110 malattia da accumulo lipidico neutro, sindrome di Conradi-Hünermann-Happle, sindrome di Hays-Wells e displasia ectodermica115 può anche occasionalmente presentarsi come collodio bambino. La membrana scompare spontaneamente nel 10% al 24% dei neonati, per lasciare il posto a una pelle completamente normale.110.116 In passato, questi casi sono stati descritti come LI del neonato, 117 ma non sono indicati come SHCB.118 Alcuni autori hanno suggerito il termine auto-miglioramento collodio ittiosi perché molti di questi pazienti, quando riesaminato più tardi durante l’infanzia o come adulti, hanno un grado variabile di anidrosi e intolleranza al calore e lievi segni di ittiosi, come xerosi e desquamazione fine, in particolare nelle ascelle e collo.78
Né la microscopia ottica né le indagini ultrastrutturali di collodion baby sono specifiche. È quindi preferibile ritardare la biopsia cutanea fino allo sviluppo del fenotipo definitivo.
Mutazioni nei geni TGM1,7,119ALOXE3,78 e ALOX12B23,78,79 sono state identificate in pazienti con SHCB. Le mutazioni di ALOX12B sono le più comuni. In una serie di 15 pazienti scandinavi con SHCB, il 67% aveva mutazioni nel gene ALOX12B, il 25% nel gene ALOXE3 e l ‘ 8,3% nel gene TGM1.78 Mutazioni non sono state trovate in alcuni pazienti, e quindi altri geni sono anche suscettibili di essere implicati. C’è stata speculazione che queste mutazioni riducono l’attività enzimatica nell’utero ma non dopo la nascita.7 Nell’utero, dove la pressione idrostatica è alta, la chelazione mediante acqua converte l’enzima mutato in una conformazione inattiva. Dopo la nascita, quando la pressione diminuisce, l’enzima ritorna alla sua forma attiva e la sua attività aumenta sufficientemente da mantenere un fenotipo normale o minimamente interessato.7
Collodion Baby auto-guaritore acrale
Sebbene collodion baby colpisca tutto il corpo, sono stati riportati casi limitati alle regioni acrali. Nel 1952, Finlay et al.120 ha riportato un caso di membrana di collodio che ha interessato solo le mani e i piedi e che ha seguito un corso di auto-guarigione. Recentemente, è stato riportato un nuovo caso di SHCB acrale in associazione con mutazioni del gene TGM1.8 Non è noto perché queste lesioni siano limitate alle regioni acrali, sebbene possano essere in funzione fattori associati alla regolazione sito-dipendente dell’attività enzimatica.8
Costume da bagno Ittiosi
Costume da bagno ittiosi è stato segnalato per la prima volta come variante ARCI indipendente nel 2005, anche se i casi di ittiosi con una distribuzione particolare erano stati segnalati in precedenza.121-123 È stato rilevato principalmente in pazienti di origine sudafricana, 9 sebbene sia stato segnalato anche in individui provenienti dall’Europa e dai paesi del Mediterraneo.124 Alla nascita, i pazienti hanno una membrana di collodio generalizzata che poi getta per lasciare la distribuzione caratteristica del ridimensionamento. Il tronco, la regione prossimale delle braccia, comprese le ascelle, il collo e il cuoio capelluto sono generalmente colpiti, mentre la parte centrale del viso, gli arti e la regione surrenale sono solitamente risparmiati.9 Le squame sono grandi, lamellari e di colore scuro. La desquamazione più fine può verificarsi nelle fosse poplitee e antecubitali.124.125 I palmi delle mani e le piante dei piedi presentano una lieve ipercheratosi diffusa, mentre il dorso delle mani e dei piedi non mostra alcun coinvolgimento.
Lo studio istopatologico della pelle colpita mostra una marcata ipercheratosi senza paracheratosi, normali strati granulari, acantosi lieve o moderata e un lieve infiltrato linfocitario nel derma superiore.9 Osservazioni di microscopia elettronica sono coerenti con ittiosi congenita di tipo 2 nella maggior parte dei casi. La pelle non coinvolta non mostra alcun risultato anormale.124.125 Nella pelle sana, l’attività della TGase 1 è leggermente ridotta e di solito localizzata nelle aree pericellulari. Nella pelle coinvolta, l’attività enzimatica è residua e anormalmente localizzata nel citoplasma.124
Sono state rilevate mutazioni nel gene TGM1 in tutti i pazienti con ittiosi da bagno studiati fino ad oggi.119,124–126 La mutazione più comune è p.Arg315Leu, che è stata identificata nella maggior parte dei pazienti sudafricani e potrebbe essere una mutazione fondante. Oji et al.124 ha suggerito che la temperatura cutanea potrebbe svolgere un ruolo nello sviluppo di queste manifestazioni. Utilizzando la termografia digitale, gli autori hanno mostrato una forte correlazione tra temperatura corporea e desquamazione, con le aree più calde del corpo che sono quelle più colpite. Aufenvenne et al.127 ha mostrato una diminuzione della temperatura ottimale per l’attività TGase 1 in pazienti con ittiosi da bagno. Questa diminuzione non è stata osservata nei controlli sani o nei pazienti con LI generalizzato. Questa diminuzione della temperatura spiegherebbe il fenotipo di questi pazienti. La temperatura ottimale è 37 ° C per l’enzima normale ma 31°C per l’enzima mutato.
Trattamento
L’obiettivo primario del trattamento nell’ittiosi è eliminare la desquamazione e ridurre la xerosi senza causare eccessiva irritazione (Tabella 3). Prima di decidere il trattamento, devono essere presi in considerazione aspetti quali età e sesso del paziente, tipo e gravità della malattia, estensione e sito delle lesioni.128
Strategia terapeutica nelle ittiosi congenite autosomiche recessive.
| Strategia terapeutica per le ittiosi congenite autosomiche recessive | |
| Bagno ed eliminazione meccanica delle squame | Bagno con bicarbonato di sodio o amido di frumento, amido di mais o amido di riso; la rimozione meccanica delle scale (1 o 2 volte al giorno) |
| trattamento Topico (sequenziale) | contenente Urea moisturizersKeratinolytics con il glicole glycolCombined keratinolytics (glicole propilenico, alfa-idrossi acidi, o urea)Keratinolytics combinato con salicilico acidTopical retinoidsIn neonati e bambini piccoli, applicare un veicolo senza principi attivi. Evitare di urea, acido salicilico e acido lattico a causa del rischio di assorbimento sistemico |
| il trattamento Orale | retinoidi Orali (acitretina o isotretinoin) |
| Altre misure | Follow-up di ectropion con il ophthalmologistRegular pulizia dell’orecchio esterno dall’orecchio-gola-naso specialistPhysiotherapy per prevenire le contratture.Evitare attività faticose in un ambiente ad alta temperatureHydrotherapy |
Balneazione ed eliminazione meccanica delle scale
Il bagno quotidiano è raccomandato per i pazienti con ARCI per eliminare meccanicamente scale e tracce di idratante. Questo è più facile se il paziente è immerso in acqua per 15-30 minuti. Alcuni autori raccomandano di aggiungere bicarbonato di sodio al bagno per denaturalizzare le cheratine e rendere l’acqua alcalina, facilitando così l’eliminazione delle squame.129 Altri prodotti che possono essere aggiunti includono amido di frumento, amido di mais o amido di riso. Gli oli da bagno non sono appropriati in quanto possono portare ad occlusione con conseguente rischio di proliferazione batterica e peggioramento della termoregolazione.
Trattamento topico
Idratanti e agenti cheratolitici topici sono di solito la prima opzione terapeutica. Migliorano la funzione della barriera cutanea e facilitano la desquamazione. Possono verificarsi lievi effetti avversi locali, come prurito transitorio, irritazione o sensazione di bruciore.
Cloruro di sodio, urea, acetato di vitamina E, glicerolo e vaselina possono essere utilizzati come idratanti e lubrificanti. Nei pazienti con desquamazione spessa e ipercheratosi marcata, possono essere aggiunti 1 o più agenti cheratolitici,come α-idrossi acidi (acido lattico e glicolico), 130 acido salicilico,N-acetilcisteina,131-133 urea (>5%), 134 e glicole propilenico. Vengono anche utilizzati modulatori della differenziazione dei cheratinociti. Questi includono retinoidi topici (tretinoina, adapalene, tazarotene),135,136 calcipotriolo,137 e dexpantenolo.I retinoidi topici spesso causano irritazione e piccole fessure molto dolorose.137 Inoltre, vi è il rischio di assorbimento e teratogenicità nelle donne fertili se vengono utilizzate troppo estensivamente.138 Per migliorare l’efficacia di cheratolitici e idratanti, la medicazione occlusiva può essere applicata in aree specifiche refrattarie al trattamento.139 Un effetto additivo o sinergico può anche essere ottenuto combinando 2 o più agenti cheratolitici o idratanti.140-142 Il trattamento deve essere ottimizzato per ogni individuo, data la natura altamente variabile della condizione e la sensibilità della pelle e le differenze nella risposta a ciascun trattamento. Il processo di ottimizzazione può essere aiutato trattando un lato del corpo in modo diverso dall’altro per consentire confronti. I neonati e i bambini piccoli devono essere trattati con un veicolo senza sostanze attive poiché la pelle è molto fine e sensibile e la maggior parte dei cheratolitici non è tollerata. Inoltre, il rischio di assorbimento percutaneo di prodotti topici come urea, acido salicilico e acido lattico è maggiore.143-145
Trattamento sistemico
I retinoidi orali hanno effetti cheratolitici che aiutano ad eliminare le squame e prevenire un’eccessiva ipercheratosi. Sia l’isotretinoina che i retinoidi aromatici (acitretina ed etretinato) si sono dimostrati efficaci nel trattamento di ARCIs.128.146.147 Acitretina ad una dose da 0,5 a 1 mg/kg / d è il farmaco più utilizzato, specialmente nei pazienti con LI.148 Pazienti con CIE possono avere una risposta più completa ea dosi più basse.
I principali effetti avversi sono disturbi mucocutanei, teratogenicità, disturbi muscoloscheletrici e profilo lipidico anormale e aumento delle transaminasi.149-152 Per quanto riguarda la teratogenicità, nel caso di etretinato e acitretina, i farmaci devono essere evitati durante la gravidanza e le pazienti devono evitare di rimanere incinte per 3 anni dopo l’interruzione del trattamento.151 L’isotretinoina ha un’emivita più breve ed è completamente eliminata dall’organismo dopo 1 mese e quindi può essere l’opzione preferita nelle donne che desiderano rimanere incinte.128
Il monitoraggio del trattamento deve includere un lavoro di laboratorio con un test di funzionalità epatica e un profilo lipidico prima di iniziare il trattamento, quindi a 1 mese e ogni 3 mesi dall’inizio del trattamento. Nelle donne fertili, un test di gravidanza deve essere eseguito nelle 2 settimane prima di iniziare il trattamento e una misura contraccettiva efficace deve essere utilizzata dalle 4 settimane prima del trattamento fino ai 3 anni successivi (nel caso di acitretina). Quando è necessario un trattamento prolungato con retinoidi, la crescita e lo sviluppo osseo devono essere monitorati. Alcuni autori suggeriscono di eseguire uno studio osseo prima del trattamento seguito da un esame annuale.151 Le recenti linee guida sconsigliano di eseguire la radiografia di routine a causa dei possibili effetti dannosi.152 Invece, si raccomandano studi radiografici selettivi in pazienti con dolore osseo atipico.152
Un’alternativa al trattamento sistemico dei retinoidi è l’uso di farmaci noti come agenti bloccanti del metabolismo dell’acido retinoico, che aumentano i livelli endogeni dell’acido retinoico. Uno di questi farmaci è il liarozolo, a cui è stato concesso lo status di orfano per il trattamento di LI, CIE e HI dall’Agenzia europea per i medicinali e dalla Food and Drug Administration statunitense.153-155 Questo farmaco ha dimostrato di essere più efficace dell’acitretina negli studi clinici ed è anche meglio tollerato e ha un profilo farmacocinetico migliore.154
Altre cure mediche
Nei pazienti con ectropion, l’applicazione di lacrime artificiali e lubrificanti per gli occhi e l’idratazione della pelle del viso e delle guance in particolare possono ridurre la retrazione palpebrale. La correzione chirurgica è un’opzione valida nei casi più gravi, ma di solito deve essere ripetuta alcuni anni dopo. L’idroterapia può essere utile.156 Pazienti devono essere avvisati di evitare un’intensa attività fisica quando la temperatura ambiente è elevata, dato che l’ipoidrosi comporta il rischio di colpo di calore e convulsioni. I retinoidi orali possono migliorare la termoregolazione.157 La fisioterapia è importante per prevenire la contrattura in flessione, in particolare nel caso dell’HI. La pulizia regolare del canale uditivo esterno da parte di uno specialista orecchio-gola-naso può impedire l’accumulo di scale e quindi prevenire la perdita dell’udito.
Consulenza genetica e diagnosi prenatale
Quando a un paziente viene diagnosticata l’ittiosi, deve essere offerta una consulenza genetica appropriata in cui siano spiegate la natura del disturbo, la modalità di trasmissione e il rischio di manifestazioni future in famiglia. La diagnosi prenatale può indicare se il feto è interessato e, se questo è il caso, può essere offerta una preparazione psicologica della famiglia e problemi previsti durante la gravidanza e il parto. Ai genitori può essere data la possibilità di un aborto se non è disponibile alcun trattamento. Inoltre, se la terapia genica per queste condizioni diventasse disponibile in futuro, la diagnosi prenatale consentirebbe l’applicazione di questa terapia il prima possibile.
Per più di 20 anni, la diagnosi prenatale è stata eseguita prelevando un campione bioptico di pelle fetale e studiandolo mediante microscopia ottica, microscopia elettronica o immunoistochimica.158.159 Questa procedura invasiva poteva essere eseguita solo nelle fasi tardive della gravidanza, tra le settimane 15 e 23 della gestazione, ed era associata a un rischio dall ‘ 1% al 3% di perdere il feto.160.161 L’identificazione dei meccanismi molecolari delle malattie ereditarie della pelle ha permesso una diagnosi molto più precoce basata su tecniche genetiche.Il DNA fetale 102,162–164 è ottenuto mediante amniocentesi eseguita tra le settimane 15 e 20 o mediante campionamento dei villi coriali tra le settimane 10 e 12. Il rischio di perdita fetale con queste tecniche è inferiore tra 0,5% e 1%.165 Altri metodi non invasivi in fase di sviluppo sono l’analisi del DNA delle cellule fetali e del DNA fetale libero nella circolazione materna166, nonché l’uso di ultrasuoni tridimensionali.167,168
La diagnosi genetica preimpianto potrebbe essere possibile anche nelle tecniche di fecondazione in vitro, in modo tale che solo le uova fecondate prive della mutazione vengano impiantate nell’utero, evitando così la necessità di aborto nella maggior parte dei casi.169
Strategie future per il trattamento genetico dell’ittiosi
Sebbene siano stati compiuti importanti progressi nella diagnosi genetica dell’ittiosi, vengono perseguite nuove strategie anche per queste malattie.170 La pelle è l’organo più accessibile per le terapie di trasferimento genico, e quindi tali tecniche sono minimamente invasive.171 Tuttavia, la pelle ha anche caratteristiche immunologiche uniche che non favoriscono l’espressione a lungo termine di un prodotto transgenico.172 In LI, un processo di trasferimento genico ex vivo è riuscito a ripristinare la normale espressione di TGM1 e correggere il fenotipo della pelle trapiantata sul retro di topi immunosoppressi.173.174 Recentemente, è stato anche recuperato il fenotipo dei cheratinociti coltivati da pazienti con HI a causa di mutazioni nel gene ABCA12.3
Conflitti di interesse
Gli autori dichiarano di non avere conflitti di interesse.