Frontiers in Neurology
Introduzione
Neuroacantocitosi è un termine superordinato per un gruppo di sindromi rare caratterizzate da sintomi neurologici in combinazione con globuli rossi deformati appuntiti (acantociti). Questo rapporto si concentra sulla Corea-acantocitosi (ChAc), che è una malattia orfana con circa 1.000 individui affetti in tutto il mondo causati da mutazioni autosomiche recessive nel gene VPS13A (vacuolar protein sorting 13 homolog A) sul cromosoma 9q (1). La malattia ha un decorso progressivo, le cause di aumento della mortalità possono essere diminuite funzioni motorie correlate a condizioni di rischio come la disfagia, ma sono state descritte anche morti improvvise e inaspettate (2, 3). Finora, non esiste un trattamento causale.
La coesistenza sospetta di acantocitosi e disturbi del movimento è stata segnalata per la prima volta negli 1970 da Levine e Critchley (4, 5) e da allora è stata accettata come la caratteristica prominente della malattia. Tuttavia, con il nostro case report descriviamo un raro fenotipo clinico di ChAc privo di evidenti disturbi del movimento, suggerendo una più ampia varietà di presentazioni cliniche. Gli strumenti diagnostici devono affrontare queste sfide per stabilire la diagnosi corretta, qui suggeriamo il neuroimaging molecolare come metodo chiave.
Case Report
Il paziente con indice maschile si è presentato per la prima volta a 25 anni di età con due crisi tonico-cloniche bilaterali non provocate. Non c’era alcuna storia medica. La risonanza magnetica cerebrale e l’EEG a quell’età erano normali e non è stato avviato alcun trattamento a causa della riluttanza del paziente e di eventi rari. Tuttavia, il paziente aveva convulsioni in corso che ora si presentano come epilessia parziale. Ha descritto i sentimenti ricorrenti di vertigini improvvise che sono state diagnosticate come un’aura vertiginosa. Inoltre, ha avuto crisi discognitive in cui ha mostrato (a) mancanza di risposta e recitazione di preghiere turche, (b) automatismi orali, o (c) giocherellare con le mani e pronunciare suoni—tutti in parte evolvendo in crisi tonico-cloniche. Video-EEG ora ha mostrato complessi spike-onda locali sia nel lobo temporale destro e sinistro. In accordo con la semiologia del sequestro, è stata fatta la diagnosi di epilessia bilaterale del lobo temporale e sono stati avviati i farmaci con levetiracetam.
All’età di 31 anni le convulsioni non sono state del tutto soppresse, come parte di ulteriori diagnosi di epilessia del lobo temporale, il paziente è stato sottoposto a un FDG-PET (Figura 1) che ha mostrato un ipometabolismo mesiotemporale bilaterale, in linea con l’origine delle crisi mesiotemporali. Tuttavia, c’era anche un marcato, altamente insolito ipometabolismo striatale che ha sollevato il sospetto di un disturbo del movimento neurodegenerativo. Di conseguenza, è stata avviata un’ampia valutazione di follow-up (cfr.Figura 2). È stato eseguito un FP-CIT-SPECT che ha prodotto una perdita moderatamente bilaterale della disponibilità del trasportatore striatale della dopamina, in linea con un declino dell’integrità nigrostriatale (Figura 1). Una risonanza magnetica ad alta risoluzione ha ora presentato atrofia bilaterale del nucleo caudato (Figura 3). L’esame neurologico ha mostrato un discreto pattern di andatura legata e ipomimia, ma nessun altro affetto del sistema motorio extrapiramidale e nessun sintomo bulbare come distonia alimentare. A parte il basso stato riflesso sugli arti inferiori, per il quale gli studi sulla conduzione nervosa hanno confermato una polineuropatia assonale sensomotoria, l’esame neurologico è stato normale. I sintomi neuropsicologici includevano aspetti di un disturbo d’ansia e il paziente riportava difficoltà con perdita di memoria a breve termine. I test neuropsicologici tuttavia non hanno confermato il malfunzionamento mnestico manifesto, ma piuttosto una distrazione aspecifica, che potrebbe essere stata ulteriormente potenziata da un’insufficiente soppressione delle convulsioni al momento. I test di laboratorio sono risultati negativi per qualsiasi epilessia sintomatica (es., anticorpi autoimmuni-encefalite, anticorpi antineuronali). Il liquido cerebrospinale non ha mostrato segni di infiammazione ma un moderato aumento proteico (765 mg/l). L’analisi delle ammine biogeniche nel liquido cerebrospinale ha rivelato un aumento della glutammina che abbiamo associato a recenti convulsioni. Nel sangue periferico è stato trovato lo 0,4% degli acantociti. Il test sierologico per la malattia di Wilson è risultato negativo. Notevole era un aumento persistente della creatin chinasi (intervallo: 1.000–3.000 U/l) che ha portato alla sospetta diagnosi di un disturbo mitocondriale. Per ulteriori indagini, è stato eseguito un test del lattato ischemico che ha mostrato risultati normali. È stata eseguita una biopsia muscolare del M. gastrocnemicus, ma l’analisi della funzione mitocondriale e della catena respiratoria era normale.
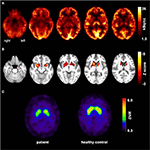
Figura 1. Risultati di studi di imaging molecolare. Le immagini transassiali FDG-PET (A) hanno mostrato non solo un lieve ipometabolismo mesiotemporale bilaterale (in linea con l’origine delle crisi mesiotemporali), ma anche un marcato ipometabolismo striatale che è risultato altamente significativo rispetto ai controlli sani . Un ulteriore esame FP-CIT-SPECT (C) ha anche rivelato una moderata perdita di trasportatori di dopamina striatale, assistendo a un declino dell’integrità nigrostriatale (un controllo sano abbinato all’età è mostrato per il confronto; DVR, rapporto del volume di distribuzione).

Figura 2. Decorso clinico e diagnostico del paziente indice.
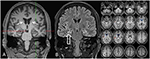
Figura 3. La risonanza magnetica mostra una discreta atrofia bilaterale della testa caudata e un ippocampo destro più piccolo e iperintenso che indica la sclerosi dell’ippocampo destro . L’ippocampo sinistro è un po ‘ iperintenso ma non atrofico. L’atrofia della testa caudata bilaterale è confermata con l’analisi CVR, in cui i colori blu mostrano una diminuzione dei colori grigi e rossi un aumento del volume del liquido cerebrospinale, rispettivamente (C).
La storia familiare ha rivelato una consanguineità dei suoi genitori (cugini di primo grado), che a loro volta non avevano una storia medica significativa. Dei suoi sei fratelli, due (di età compresa tra i 20 e i 30 anni) avevano già sofferto di convulsioni generali (vedi Figura 4). Le cartelle cliniche dei suoi fratelli non erano a nostra disposizione.
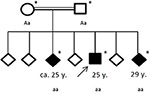
Figura 4. Pedigree della famiglia colpita, con l’età del primo attacco epilettico in anni (y). Freccia: indice di brevetto. Asterisco: test genetici eseguiti. Aa, eterozigoti; aa, omozigoti per c. 4326 T > A (p. Tyr1442*).
Sotto l’ipotesi di un disturbo del movimento neurodegenerativo ereditario, il paziente e la sua famiglia sono stati sottoposti a test genetici. Il sequenziamento dell’esoma ha rivelato una nuova mutazione omozigote troncante c. 4326 T > A (p. Tyr1442*) nel gene ChAc VPS13A nei tre fratelli affetti. I genitori consanguinei sono stati entrambi rilevati eterozigoti. Non sono state identificate altre varianti o mutazioni nel sequenziamento dell’esoma.
Infatti, in un follow-up all’età di 33 anni, il paziente indice presentava ancora solo sintomi extrapiramidali marginali e nessuno era presente in nessuna delle sue sorelle. Ora ha dato l’impressione di una leggera rigidità solo sotto l’adescamento sull’arto superiore destro e una lieve bradicinesia degli arti superiori. In terapia con levetiracetam e lacosamide, il paziente è risultato privo di crisi epilettiche.
Discussione
Con il presente caso, dimostriamo che ChAc può manifestarsi clinicamente con epilessia del lobo temporale priva di sintomi motori significativi a causa di una nuova mutazione corrispondente nel gene VPS13A.
Il decorso della malattia è tipicamente caratterizzato da un disturbo progressivo del movimento (tra cui corea, distonia, parkinsonismo), alterazioni cognitive e psichiatriche e sintomi miopatici con marcatori sierologici di acantocitosi e iperckemia. Le convulsioni sono state precedentemente notate come un sintomo di ChAc (6), ma i disturbi del movimento sono ancora considerati il sintomo chiave. L’evidenza sorge che l’epilessia, e più specificamente l’epilessia che ha origine nel lobo temporale, potrebbe essere un fenotipo sottovalutato di ChAc. Peluso et al. ha riportato un paziente simile al nostro, che, anche all’età di 46 anni, non ha mostrato disturbi del movimento mentre presentava risultati di neuroimaging indicativi di coinvolgimento dei gangli della base (7). In accordo con ciò, Scheid et al. descritto tre pazienti che hanno sofferto di epilessia del lobo temporale mesiale e sclerosi (sulla base di studi MRI) come il sintomo predominante e sono stati diagnosticati con ChAc (8). Inoltre, Mente et al. recentemente fornito i risultati dell’autopsia di un paziente ChAc che ha mostrato non solo l’atrofia dei gangli della base, ma anche la sclerosi ippocampale (9). In linea con il nostro caso, il coinvolgimento bilaterale dell’ippocampo sembra essere una scoperta comune (10, 11). L’esatta relazione tra convulsioni e sclerosi è, tuttavia, ancora una questione di gallina o uovo. Il ruolo correlato della coreina nello sviluppo strutturale dell’ippocampo non è ancora completamente compreso. È interessante notare che in un modello murino di ChAc è stato osservato un cambiamento strutturale, poiché l’espressione proteica dell’ippocampo del recettore GABA (A) gamma 2 e la sua proteina di ancoraggio chorein sono state aumentate (12).
Nella routine clinica, questo fenotipo può ancora essere difficile nel trovare la diagnosi corretta. Il primo suggerimento cruciale verso una malattia neurodegenerativa nel nostro paziente è stato un declino prominente nel metabolismo del glucosio striatale e un moderato declino dell’integrità nigrostriatale su FDG-PET e FP-CIT-SPECT, rispettivamente. La piccola raccolta di risultati di imaging funzionale, basata sulla descrizione della serie di casi, mostra che le alterazioni del metabolismo e la disfunzione dopaminergica si verificano principalmente nel nucleo caudato e nel putamen (13). La risonanza magnetica cerebrale ha rivelato l’atrofia del nucleo caudato durante il decorso della malattia, che è stata descritta come una scoperta tipica in ChAc (14). Di conseguenza, le caratteristiche neurologiche distinte di solito includono corea, distonia alimentare, discinesia orofaciolingue e tic, insieme ad altri sintomi di affezione dei gangli basali come parkinsonismo e distonia (3). Si è tentati di ipotizzare che i cambiamenti striatali osservati nel nostro paziente fossero ancora al di sotto della soglia di causare sintomi (cioè, risultati di imaging prodromico), che alla fine possono verificarsi man mano che la malattia progredisce. Incoraggiamo quindi l’imaging strutturale e molecolare come strumento diagnostico sensibile in questa malattia orfana.
In ChAc, ci sono descrizioni della conta degli acantociti tra il 5 e il 50% (3), ma ci sono alcuni casi che dimostrano che gli acantociti possono comparire solo in ritardo nel corso della malattia (15), potrebbero essere molto bassi o addirittura completamente assenti (16). Pertanto, gli acantociti non dovrebbero essere considerati obbligatori per fare la diagnosi. Inoltre l’epilessia potrebbe ritardare la diagnosi in quanto nasconde altri sintomi tipici di ChAc. In particolare, i sintomi psichiatrici come i cambiamenti di personalità e il deterioramento cognitivo, che sono molto comuni, possono essere interpretati erroneamente e attribuiti a effetti collaterali correlati al farmaco (ad esempio, di levetiracetam) o correlati alle convulsioni. L’iperckemia secondaria è veduta dopo le crisi cloniche toniche generali e un’elevazione persistente potrebbe essere trascurata.
Il nostro caso evidenzia anche il vantaggio cruciale del sequenziamento dell’esoma per stabilire una diagnosi corretta, specialmente nelle malattie rare o quando i sintomi sono vaghi. La famiglia dei geni VPS13 (A–D) dei mammiferi è di crescente interesse e sono state identificate mutazioni in altre malattie neurologiche come il morbo di Parkinson autosomico recessivo o l’atassia spinocerebellare autosomica recessiva (17). Mutazioni recessive in VPS13D causano disturbi del movimento dell’infanzia (18). La fisiopatologia sottostante di ChAc non è ancora completamente decifrata, ma è stato dimostrato che VPS13A codifica la proteina choreina che è espressa ubiquitamente nel cervello e in un vasto numero di altri tessuti (19). Gli studi rivelano che partecipa alle vie di segnalazione che regolano l’architettura citoscheletrica, l’esocitosi e la sopravvivenza cellulare (20). Un’altra mutazione che è stata descritta per presentare un fenotipo dominato da convulsioni in 9 pazienti con ChAc è la mutazione c.2343del nel gene VPS13A (21). È ragionevole supporre che mutazioni specifiche nello stesso gene provochino una certa aberrazione della coreina che causa un fenotipo unico, ad esempio interessando diverse aree del cervello. Tuttavia, la fisiopatologia sottostante è ancora solo speculativa e un’ulteriore documentazione delle mutazioni causali sarà di interesse. Mostriamo qui che la mutazione troncante c. 4326 T > A (p. Tyr1442*), che non è stata descritta prima, risulta in un fenotipo simile con epilessia predominante in tutti i membri della famiglia colpiti.
Osservazioni conclusive
È probabile che l’epilessia (in particolare l’epilessia del lobo temporale bilaterale) sia un fenotipo predominante di ChAc. Pertanto, una ricerca di ulteriori bandiere rosse (come iperckemia, storia familiare, polineuropatia) deve essere eseguita con attenzione. In casi sospetti ricerca di acantociti nel sangue e il cervello MRI dovrebbe essere aggiunto come questi strumenti sono ampiamente disponibili. In caso di dubbio la diagnostica deve essere integrata con FDG-PET e/o FP-CIT-SPECT poiché sono sensibili nel rilevare il coinvolgimento striatale. Nelle epilessie autosomiche recessive con risultati indicativi per una malattia neurodegenerativa VPS13A test genico singolo o chorein Western blot dovrebbe quindi essere utilizzato per stabilire la diagnosi corretta. Quest’ultimo dovrebbe anche essere considerato in altri disturbi neurodegenerativi senza eziologia geneticamente definita.
Dichiarazione etica
Il consenso informato scritto è stato ottenuto dal paziente per la partecipazione allo studio. Il consenso informato scritto è stato ottenuto dal paziente per la pubblicazione di questo rapporto di caso.
Contributi dell’autore
JW, MR e SK hanno partecipato alla gestione del paziente. JW ha scritto la prima bozza del manoscritto. LF, HU, e PM figure preparate. Tutti gli autori hanno contribuito alla revisione del manoscritto, hanno letto e approvato la versione presentata.
Dichiarazione sul conflitto di interessi
HU è azionista di Veobrain Gmbh, uno spin-off del Centro medico universitario di Friburgo.
I restanti autori dichiarano che la ricerca è stata condotta in assenza di rapporti commerciali o finanziari che potrebbero essere interpretati come un potenziale conflitto di interessi.
1. Il nostro sito utilizza cookie tecnici e di terze parti. La neuropsichiatria delle sindromi da neuroacantocitosi. Il sito utilizza cookie tecnici e di terze parti. doi: 10.1016 / j. neubiorev.2011.01.001
PubMed Abstract / CrossRef Testo completo / Google Scholar
2. Walker RH. Gestione delle sindromi da neuroacantocitosi. Tremore Altro Hyperkinet. Mov. (2015) 5:346. doi: 10.7916 / D8W66K48
PubMed Abstract / CrossRef Testo completo / Google Scholar
3. Velayos Baeza A, Dobson-Stone C, Rampoldi L, Bader B, Walker RH, Danek A, et al. Corea-Acantocitosi. GeneReviews®. Seattle, WA: Università di Washington (1993).
4. Critchley EM, Clark DB, Wikler A. Acantocitosi e disturbo neurologico senza betalipoproteinemia. Arch Neurol. (1968) 18:134–40.
PubMed Abstract
5. Levine IM, Estes JW, Looney JM. Malattia neurologica ereditaria con acantocitosi. Una nuova sindrome. Arch Neurol. (1968) 19:403–9.
PubMed Abstract / Google Scholar
6. Il sito utilizza cookie tecnici e di terze parti per migliorare la tua esperienza di navigazione. Neuroacantocitosi. uno studio clinico, ematologico e patologico di 19 casi. Brain (1991) 114(Pt 1A):13-49.
PubMed Abstract / Google Scholar
7. Peluso S, Bilo L, Esposito M, Antenora A, Rosa ADR, Pappata S, et al. Corea-acantocitosi senza corea: espansione del fenotipo clinico. Parkinson Relat Disturbo. (2017) 41:124–6. doi: 10.2016 / j. parkreldis.2017.05.013
PubMed Abstract / CrossRef Testo completo / Google Scholar
8. Scheid R, Bader B, Ott DV, Merkenschlager A, Danek A. Sviluppo dell’epilessia del lobo temporale mesiale in corea-acantocitosi. Neurologia (2009) 73:1419-22. doi: 10.1212 / WNL.0b013e3181bd80d4
PubMed Abstract / CrossRef Testo completo / Google Scholar
9. Mente K, Kim SA, Grunseich C, Hefti MM, Crary JF, Danek A, et al. Sclerosi ippocampale e epilessia del lobo temporale mesiale in corea-acantocitosi: un caso con valutazione clinica, patologica e genetica. Neuropatolo Appl Neurobiol. (2017) 43:542–6. doi: 10.1111 / nan.12403
PubMed Abstract / CrossRef Testo completo / Google Scholar
10. Bader B, Vollmar C, Ackl N, Ebert A,la Fougère C, Noachtar S, et al. Epilessia bilaterale del lobo temporale confermata con EEG intracranico in corea-acantocitosi. Sequestro (2011) 20:340-2. doi: 10.1016 / j.sequestro.2010.12.007
PubMed Abstract / CrossRef Testo completo / Google Scholar
11. Marson AM, Bucciantini E, Gentile E, Geda C. Neuroacantocitosi: risultati clinici, radiologici e neurofisiologici in una famiglia italiana. Neurol Sci. (2003) 24:188–9. doi: 10.1007 / s10072-003-0123-1
PubMed Abstract / CrossRef Testo completo / Google Scholar
12. Kura Y, Nakamura M, Ichiba M, Matsuda M, Mizuno E, Kato M, et al. La carenza di chorein conduce a upregulation del gephyrin e del ricevitore di GABA(A). Biochem Biophys Res Commun. (2006) 351:438–42. doi: 10.1016 / j. bbrc.2006.10.070
PubMed Abstract / CrossRef Testo completo / Google Scholar
13. Ehrlich DJ, Walker RH. Neuroimaging funzionale e corea: una revisione sistematica. J Clin Mov Disordine. (2017) 4:8. doi: 10.1186 / s40734-017-0056-0
PubMed Abstract / CrossRef Testo completo / Google Scholar
14. Connolly BS, Hazrati L-N, Lang AE. Scoperte neuropatologiche in corea-acantocitosi: nuove intuizioni sui meccanismi alla base del parkinsonismo e delle convulsioni. Acta Neuropatolo. (2014) 127:613–5. doi: 10.1007 / s00401-013-1241-3
PubMed Abstract / CrossRef Testo completo / Google Scholar
15. Sorrentino G, De Renzo A, Miniello S, Nori O, Bonavita V. Comparsa tardiva di acantociti durante il corso della corea-acantocitosi. J Neurol Sci. (1999) 163:175–8.
PubMed Abstract / Google Scholar
16. Bayreuther C, Borg M, Ferrero-Vacher C, Chaussenot A, Lebrun C. Choréo-acanthocytose sans acanthocytes. Revue Neurol. (2010) 166:100–3. doi: 10.1016 / j.neurol.2009.03.005
CrossRef Testo completo / Google Scholar
17. Lesage S, Drouet V, Majounie E, Deramecourt V, Jacoupy M, Nicolas A, et al. La perdita della funzione VPS13C nel parkinsonismo autosomico-recessivo causa disfunzione mitocondriale e aumenta la mitofagia PINK1/Parkin-dipendente. Sono J Hum Genet. (2016) 98:500–13. doi: 10.1016 / j.ajhg.2016.01.014
PubMed Abstract / CrossRef Testo completo / Google Scholar
18. Gauthier J, Meijer IA, Lessel D, Mencacci NE,Krainc D, Hempel M, et al. Mutazioni recessive in > VPS13D causano disturbi del movimento ad esordio infantile. Ann Neurol. (2018)83: Io6. doi: 10.1002 / ana.25204
PubMed Abstract / CrossRef Testo completo / Google Scholar
19. Kura Y, Nakamura M, Ichiba M, Matsuda M, Mizuno E, Kato M, et al. Distribuzione in vivo e localizzazione di chorein. Biochem Biophys Res Commun. (2007) 353:431–5. doi: 10.1016 / j. bbrc.2006.12.059
PubMed Abstract / CrossRef Testo completo / Google Scholar
20. Lang F, Pelzl L, Schöls L, Hermann A, Föller M, Schäffer TE, et al. Neuroni, eritrociti e oltre-le diverse funzioni della coreina. Neurosignals (2017) 25:117-26. doi: 10.1159/000485457
PubMed Abstract / CrossRef Testo completo / Google Scholar
21. Il suo nome deriva dal greco antico, che significa “albero di natale”, “albero di natale”, “albero di natale”, “albero di natale”, “albero di natale”, “albero di natale”, “albero di natale”, “albero di natale” e “albero di natale”. Convulsioni come presentazione e sintomo prominente in corea-acantocitosi con mutazione del gene c. 2343del VPS13A. Epilepsia (2016) 57:549-56. doi: 10.1111 / epi.13318
PubMed Abstract | CrossRef Full Text | Google Scholar