Frontiers in Pediatrics
- Introduzione
- Sindrome del QT lungo
- LQTS1
- LQTS2
- LQTS3
- LQTS4
- LQTS5
- LQTS6
- LQTS7
- LQTS8
- LQTS9 e LQTS10
- LQTS11
- LQTS12
- LQTS13
- LQTS acquisito
- Caratteristiche elettrocardiografiche nelle tre forme comuni di sindrome del QT lungo
- Genotipo-fenotipo
- Gestione clinica di LQTS
- LQT: Prospettiva saudita
- Dichiarazione sul conflitto di interessi
- Ringraziamenti
Introduzione
Le aritmie cardiache familiari o ereditarie comprendono percentuali significative di aritmie e anche causali alla morte cardiaca improvvisa (SCD) (1, 2). Negli ultimi due decenni, scienziati e medici hanno fornito enormi sforzi per svelare i meccanismi intricati e complessi delle aritmie familiari congenite (1-8). Per comprendere il meccanismo dell’aritmogenesi, è necessario conoscere le basi della struttura cellulare cardiaca e le loro proprietà elettrofisiologiche. I miociti cardiaci sono le principali cellule funzionanti nel cuore e sono ampiamente accoppiati in modo che gli impulsi si propaghino rapidamente e in modo uniforme. I cardiomiociti sono separati l’uno dall’altro da un confine specializzato chiamato disco intercalato; proteine di giunzione gap, desmosomi cardiaci e canali ionici si trovano nel disco intercalato (9). Le giunzioni Gap sono costituite da connessine strettamente imballate che consentono lo scambio intercellulare di piccole molecole e consentono anche di fluire le correnti eccitatorie da una cellula alla cellula vicina. I desmosomi insieme alle giunzioni aderenti sono responsabili degli attacchi meccanici dei singoli cardiomiociti. Tutti questi componenti nei dischi intercalati sono sequenzialmente segregati e ogni componente esercita la sua funzione unica; la rottura di un componente influenza la funzione di altri componenti, che predispone il cuore a sviluppare aritmie (9-11). I canali ionici cardiaci sono complessi proteici che formano pori che forniscono il movimento interno ed esterno delle correnti ioniche attraverso le membrane cellulari, essenziale per la generazione e la propagazione del ritmo cardiaco. La sindrome del QT lungo (LQTS), sindrome del QT corto (SQTS), sindrome del seno malato (SSS), difetti di conduzione cardiaca (CCD), BrS, catecholaminergic tachicardia ventricolare polimorfa (CPVT), quella della ripolarizzazione precoce, sindrome di (ERS), e familiare Fibrillazione Atriale (AF) sono attualmente noti canalopatie cardiache, che potrebbe verificarsi a causa di un singolo o più difetti in geni legati al ritmo cardiaco generazione e propagazione.
In questa recensione, descriveremo esclusivamente le aritmie collegate a LQTS, la sua fisiopatologia e la gestione clinica attualmente disponibile. Siamo stati pionieri nel chiarire la patologia genetica nelle aritmie cardiache familiari in Arabia Saudita (1, 12-14), stiamo ancora conducendo indagini cardiogenetiche in Arabia Saudita, alla fine di questa recensione, discuteremo le conoscenze attualmente disponibili sui risultati genetici e clinici ottenuti da diverse famiglie saudite con storia di sincope e SCD. Aggiungeremo anche i dati recentemente pubblicati su LQTS da un team del King Faisal Specialist Hospital and Research Centre, Riyadh (15).
Sindrome del QT lungo
LQTS congenito è un disturbo ereditario definito dal prolungamento dell’intervallo QT all’elettrocardiogramma (ECG). I pazienti con tutte le forme di LQTS sono predisposti alla tachiaritmia ventricolare, alle torsioni di punta (TdP) che portano a sincope ricorrente o SCD. In molti casi, la sincope o la morte improvvisa potrebbero essere la prima e l’unica manifestazione. LQTS colpisce circa 1 persona su 2.000 in tutto il mondo (16). Il segno distintivo del LQTS è il prolungamento dell’intervallo QT sull’ECG (corretto per la frequenza cardiaca, cioè QTc). I valori normali di QTc sono 440 ms nei maschi e 450 ms nelle femmine. Nei bambini, i valori dipendenti dall’età e dal sesso sono rilevanti. Recente raccomandazione di consenso per la diagnosi LQTS sono i seguenti (17, 18):
1. LQTS viene diagnosticato:
a. In presenza di un punteggio di rischio LQTS ≥3,5 in assenza di una causa secondaria per il prolungamento del QT e/o
b. In presenza di una mutazione inequivocabilmente patogena in uno dei geni LQTS o
c. In presenza di un intervallo QT corretto per la frequenza cardiaca usando la formula di Bazett (QTc) ≥500 ms in elettrocardiogramma ripetuto a 12 conduttori e in assenza di una causa secondaria per il prolungamento del QT.
2. LQTS può essere diagnosticata in presenza di un QTc tra 480 e 499 ms in ECG ripetuti a 12 conduttori in un paziente con sincope inspiegabile in assenza di una causa secondaria di prolungamento del QT e in assenza di una mutazione patogena.
I pazienti con durata del QTc ≥500 ms hanno avuto una probabilità cumulativa significativamente più alta di manifestare la loro prima sincope rispetto ai pazienti con durata del QTc < 500 ms dalla nascita fino all’età di 20 anni (19). Tuttavia, i pazienti che hanno avuto 2, 3 e 4 episodi di sincope, il rischio di un successivo episodio di sincope era praticamente identico tra i pazienti che avevano una durata ridotta o prolungata del QTc (19). La base molecolare di LQTS è eterogenea e ad oggi sono state descritte mutazioni in 13 diversi geni causali di LQTS (1-8, 19, 20). Un difetto genetico si trova di solito nel 70% dei pazienti con LQTS in uno di questi 13 geni (1-8, 19, 20). Tra i 13 diversi tipi di LQTS attualmente noti, i più comuni sono LQTS1, LQTS2 e LQTS3, a causa di difetti nei geni del canale ionico cardiaco, KCNQ1, KCNH2 e SCN5A, rispettivamente. Il novanta per cento di tutte le mutazioni causali LQTS si trovano in questi tre geni (1-8, 19, 20). Le mutazioni nei restanti 10 geni sono rare e comprendono solo only il 10% di tutte le mutazioni LQTS attualmente note.
La sindrome del QT lungo è di solito una malattia autosomica dominante, ma, occasionalmente, mutazioni multiple in un singolo gene o in geni diversi potrebbero essere trovate nel 5-10% dei pazienti con LQTS (21, 22). I pazienti con mutazioni multiple potrebbero presentare un QTc più lungo rispetto a quelli con una singola mutazione e tali pazienti sono anche a increased 3,5 volte aumentato rischio di eventi cardiaci potenzialmente letali(21, 22)
LQTS1
Mutazioni nel gene KCNQ1 sono la forma più comune di tutti i LQTS e indicato come LQTS tipo 1 (LQTS1). Il cinquanta per cento di tutte le mutazioni causali LQTS si trovano nel gene KCNQ1 (20). KvLQT1 (chiamato anche Kv7.1) è una proteina fatta da KCNQ1 gene e la proteina forme tetrameri nel reticolo endoplasmatico all’interno delle cellule, che putativamente co-assembla con la proteina di visone (codificato da KCNE1) e sono quindi trasportato alla membrana plasmatica dei miociti cardiaci, dove si mediare una lenta attivazione di corrente che accelera la ripolarizzazione del potenziale d’azione nei tessuti cardiaci, questa corrente è conosciuto come IKs (Figura 1). Le mutazioni causali KCNQ1 di LQTS1 sono per lo più mutazioni missense e in rari casi potrebbero essere mutazioni di frameshift nella regione C-terminale (23). L’aritmia cardiaca nei portatori di mutazione KCNQ1 è innescata da pulsioni adrenergiche, ad esempio stress emotivo, sforzo fisico, immersioni, nuoto (24-26). I pazienti con mutazioni nei domini transmembrana di KvLQT1 sono a più alto rischio di eventi cardiaci correlati a LQTS e hanno una maggiore sensibilità alla stimolazione simpatica (26, 27). I pazienti con mutazioni missense sono a rischio aumentato rispetto ai pazienti con mutazioni non sensoriali o troncanti (27, 28). La variazione del 3 ‘ – UTR nel gene KCNQ1 influisce anche in modo significativo sulla suscettibilità all’aritmia, presumibilmente influenzando l’espressione del gene (29). I pazienti con aritmie dovute a mutazioni KCNQ1 rispondono abbastanza bene ai β-bloccanti, ma alcuni pazienti potrebbero ancora essere meno reattivi o addirittura resistenti a questo farmaco. In un recente articolo, Barsheshet et al. (30) ha affermato che i pazienti con mutazioni al di fuori della regione del ciclo citoplasmatico (c-loop) nel KvLQT1 sono meno sensibili ai β-bloccanti.
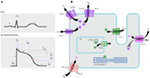
Figura 1. Correnti ioniche che contribuiscono al potenziale d’azione ventricolare (A) e rappresentazione schematica di un cardiomiocita che mostra (solo) quelle proteine coinvolte nella patogenesi delle sindromi ereditarie dell’aritmia (B). In (A), il potenziale d’azione è allineato con il suo tempo approssimativo di azione durante l’ECG. In (B), ankyrin-B, una proteina adattatore coinvolta nella sindrome del QT lungo di tipo 4, non è raffigurato.
Mutazioni eterozigoti omozigoti o composti nel gene KCNQ1, che potrebbero causare la forma recessiva della malattia, la sindrome di Jervell e Lange-Nielsen (JLNS), di tipo 1 (31), sono piuttosto rare. I pazienti affetti da JLNS soffrono anche di gravi aritmie cardiache e sordità (31, 32). I pazienti con mutazioni KCNQ1 causali a JLNS di solito non hanno alcun IKS funzionale (28, 31-33). È possibile che alcuni pazienti non possano avere sordità pur avendo mutazioni eterozigoti omozigoti o composti in KCNQ1, tali casi sono indicati come autosomica recessiva LQTS1 (13, 32). In questi pazienti, una piccola quantità di corrente IKs funzionale (<10% del totale IKs) potrebbe ancora essere presente, il che mantiene la funzione uditiva, ma i loro difetti del ritmo cardiaco sono ugualmente gravi come nei pazienti con JLNS (13, 32).
LQTS2
Questo tipo di LQTS è ugualmente prevalente come LQTS1, rappresentando il 35-40% dei pazienti con LQTS con una mutazione rilevabile (1, 20, 24, 25). KCNH2 codifica per la proteina HERG (Kv11.1), che è la subunità α del raddrizzatore ritardato ad attivazione rapida K+ corrente (IKr). Mutazioni patogene in questo gene che riduce la funzione del canale Kv11.1 prolunga la durata dell’intervallo QT (Figura 1) e sono causali a LQTS2. Ventinove per cento degli attacchi sincopali in LQTS2 si verificano durante il riposo / sonno e solo il 13% degli attacchi sincopali sono stati segnalati per verificarsi durante l’esercizio (25, 34). Rumori improvvisi, ad esempio, anelli di sveglia, campanelli, squilli telefonici, tipicamente innescano attacchi sincopali in questi pazienti (24, 34). I pazienti con mutazioni nella regione di formazione dei pori del Kv11.1 (codificato dal gene KCNH2) sono suscettibili ad alto rischio di eventi cardiaci correlati all’aritmia rispetto ai pazienti con mutazioni nella regione non dei pori (35). Nei neonati, il blocco atrioventricolare 2: 1 (AV) è preferenzialmente associato a mutazioni KCNH2 (36). Blocco AV completo complicato da LQTS sono stati trovati anche nel 17% dei pazienti adulti con una mutazione nel gene KCNH2 (37). Le mutazioni omozigoti in KCNH2 sono rare e, quando presenti, i pazienti soffrono di una forma grave di LQTS, con 2:1 Blocco AV e gravi aritmie ventricolari, durante le fasi intrauterine e dopo la nascita (11, 38-40). Oltre alle numerose mutazioni descritte nella patologia LQTS2, varianti polimorfiche comuni nel gene KCNH2 potrebbero anche modulare la gravità della malattia. Un esempio interessante è il polimorfismo K897T (SNP) in KCNH2, che è presente nel 33% della popolazione generale (41). K897T è stato segnalato per esacerbare la patogenicità delle mutazioni KCNH2 (42, 43).
LQTS3
Nav1.5 è la subunità α-formante pori del canale Na + cardiaco voltaggio-dipendente, è una proteina di membrana integrale codificata dal gene SCN5A ed è coinvolta nell’iniziazione e nella conduzione dei potenziali di azione cardiaca. I canali Na + cardiaci sono composti da una subunità α che forma pori (codificata da SCN5A) e da una o più subunità β ausiliarie.
LQTS3 è causato da mutazioni del guadagno di funzione che interrompono la rapida inattivazione della subunità α (Figura 1) e una mutazione nel gene SCN5A è stata descritta in <10% di tutti i pazienti con LQTS con una mutazione (1, 20, 24). I pazienti con LQTS3 presentano la maggior parte (39%) dei loro eventi cardiaci durante il sonno/riposo (25), e circa ∼il 13% degli eventi è stato segnalato durante l’esercizio fisico (25). In parecchi casi, una singola mutazione SCN5A è stata indicata per esercitare due o persino tre fenotipi distinti delle aritmie nella stessa famiglia, per esempio, LQTS, BrS, o CCD (44-47). I pazienti di sesso maschile con una mutazione LQTS3 potrebbero sviluppare sintomi molto prima rispetto ai pazienti di sesso femminile (48). Mutazioni SCN5A sia eterozigoti che omozigoti sono state descritte in LQTS3 con blocco AV funzionale 2: 1 (49). Recentemente abbiamo trovato una famiglia iraniana con la mutazione 1507_1509delQKP in più membri della famiglia, in cui i pazienti avevano combinato LQTS e CCD (dati non mostrati). Questa mutazione è stata riportata anche in pazienti di altri paesi, il che suggerisce che si tratta di una mutazione ricorrente e hot spot . Mutazioni con tali caratteristiche di perdita e guadagno di funzione durante le diverse fasi del potenziale d’azione sono state riportate anche nelle mutazioni 1493delK e 1795insD (44, 51).
Occasionalmente, un SNP potrebbe anche esercitare un effetto patogeno sui suoi portatori. S1103Y è una variante comune nel gene SCN5A, presente nel 13% degli afroamericani (52). I portatori con questa variante sono ad aumentato rischio di aritmie e sindrome da morte improvvisa infantile (SIDS) (52).
LQTS4
LQTS4 rappresenta la prima forma non-canale di LQTS. Una mutazione nel ANK2, una proteina adattatore, porta a sovraccarico di calcio intracellulare che contribuisce alla LQTS4 (53, 54). Oltre al prolungamento del QT, i pazienti con questa sindrome potrebbero avere bradicardia sinusale, AF parossistica e CPVT (54). L’effetto patogeno delle mutazioni ANK2 potrebbe essere da moderato a grave e le espressioni cliniche dipendono dalla gravità della mutazione.
LQTS5
Le mutazioni in KCNE1 sono associate alla LQTS5 (Figura 1) (55, 56). Le mutazioni eterozigoti di KCNE1 riducono IKs esercitando un effetto negativo dominante sul suo allele normale di accompagnamento e portano a una ripolarizzazione cardiaca ritardata (Figura 1), responsabile di un aumento del rischio di aritmie (6). I pazienti con mutazioni KCNE1 omozigoti soffrono di JLNS (tipo 2) (57, 58).
D85N è un polimorfismo nel gene KCNE1, presente in 0.7-1% della popolazione generale (41). In uno studio di Nishio et al. (59), il polimorfismo D85N è stato riscontrato più frequentemente nei pazienti con LQTS, rendendolo un genotipo di rischio nella patologia LQTS (potenzialmente solo nella popolazione asiatica). In Europa, D85N è stato riportato nel 5% dei pazienti LQTS acquisiti (ALQTS) (in una coorte di 32 pazienti), che avevano manifestato TdP (60).
LQTS6
Il gene KCNE2 codifica per il peptide 1 correlato al MinK (MiRP1), una presunta subunità β del canale cardiaco del potassio IKr (Figura 1). Mutazioni nel gene KCNE2 potrebbero anche portare a difetti nella componente ad attivazione rapida della corrente di potassio del raddrizzatore ritardato (IKr), base patologica di LQTS6 (61). Stimolo uditivo / acustico come il rumore della sveglia, il campanello ecc. potrebbe provocare attacchi sincopali nei portatori di mutazione KCNE2, simili alla mutazione KCNH2 (62).
LQTS7
Questa sindrome è anche conosciuta come sindrome di Andersen–Tawil (ATS). L’ATS è una malattia rara, manifestata da sincope occasionale e arresto cardiaco. Le caratteristiche dell’ECG includono un lieve prolungamento dell’intervallo QT, onde U anormali, ectopia ventricolare frequente, tachicardia ventricolare bidirezionale (VT) e VT polimorfico. Questa sindrome presenta anche caratteristiche extracardiache, ad esempio paralisi periodica del muscolo scheletrico e problemi di sviluppo, come palatoschisi, orecchie basse, bassa statura e difetti dello sviluppo negli arti (63). La maggior parte dei pazienti con ATS clinicamente diagnosticati ha riportato una mutazione in KCNJ2 (63). KCNJ2 codifica una subunità che forma i pori dei canali del potassio rettificanti internamente (IK1) (Figura 1) (64, 65).
LQTS8
Noto anche come sindrome di Timothy (TS), i pazienti mostrano un grave prolungamento del QT sul loro ECG, che è combinato con sindattilia, calvizie alla nascita e piccoli denti nel 100% dei casi e malformazioni strutturali cardiache meno penetranti, ritardo mentale, autismo e caratteristiche dismorfiche facciali (66). Ci sono due sottotipi: TS1 (classico) e TS2 (forma rara).
TS2 è cardiologicamente più severo di TS1 (66, 67). I pazienti con TS2 mancano anche di sindattilia (67). Mutazioni nella subunità α-1 della corrente di calcio di tipo L (CAC-L) che codifica il gene CACNA1C portano ad entrambe le forme di TS (LQTS8). Ci sono alternativamente impiombati, mutuamente esclusivi esone-8 nel gene CACNA1C, per chiarezza, sono chiamati come esone – 8 ed esone-8A. Nel cuore e nel cervello, dove il CACNA1C è prevalentemente espresso, l’esone-8 si trova in 8 80% delle trascrizioni di mRNA e l’esone-8A è presente transc 20% trascrizioni (66). La mutazione G406R nell’esone-8A è causale alla forma classica di TS (TS1) e G402S nell’esone-8 è stata riportata in severer nel TS2. Una nuova aggiunta a questa lista è la mutazione Ala1473Gly, che è stata descritta in un bambino TS con fenotipo ulteriormente espanso (68). Tutte le mutazioni erano de novo o mosaico nel genitore, e tutti risultato guadagno di funzione del canale IC-L (66-69).
LQTS9 e LQTS10
Mutazioni in CAV3 o SCN4B producono guadagno di funzione in ritardo INa, causando un fenotipo simile a LQTS3 (70-74). Sono noti come LQTS9 (associato alla mutazione CAV3) e LQTS10 (associato alla mutazione SCN4B).
Le caveole sono ben descritte da Engelman et al. (72) come “piccole caverne” nella membrana plasmatica. Sono piccole fosse non rivestite ed è considerato come il sito di importanti eventi dinamici e regolatori alla membrana plasmatica (72, 73). Le caveoline sono le principali proteine nelle caveole e la caveolina-3 (codificata dal gene CAV3) si trova specificamente nei cardiomiociti e nelle cellule muscolari scheletriche. Diversi canali ionici cardiaci sono stati specificamente segnalati per essere localizzati nelle caveole estratte da miociti cardiaci arricchiti in caveolina-3 (72, 73). Inoltre, i componenti della cascata di segnalazione del recettore β-adrenergico sono presenti anche nelle membrane arricchite da caveole (72, 73).
SCN4B codifica per NaVß4, che è una subunità β ausiliaria del canale del sodio cardiaco. Finora solo una mutazione (L179F) è stata riportata in questo gene in una famiglia messicana con più membri della famiglia affetti (74). La mutazione in questo gene è stata trovata per provocare il guadagno della funzione della corrente Nav1.5 (74).
LQTS11
Nel cuore, la regolazione simpatica della durata del potenziale d’azione cardiaco (APD) è mediata dall’attivazione del recettore β-adrenergico (β-AR), che richiede l’assemblaggio di AKAP9 (Yotiao) con la subunità α (KvLQT1) del canale IKs. La mutazione in AKAP9 causa LQTS11 (75). Ad oggi è stata riportata solo una mutazione, S1570L, in AKAP9 (75).
LQTS12
La mutazione nel gene α-1-Syntrophin (SNTN1) è causale per LQTS12. La mutazione in questo gene conduce a guadagno di funzione del canale di sodio cardiaco (Nav1.5), che è la base patologica di LQTS12 (76).
LQTS13
La subunità del canale del potassio accoppiata alla proteina G (Kir3.4) è codificata dal gene KCNJ5. Una mutazione di perdita di funzione in questo gene potrebbe causare LQTS13 (77). Finora, solo una mutazione, G387R, è stata descritta in una famiglia cinese con nove pazienti che hanno questa mutazione. L’espressione ridotta della membrana plasmatica di Kir3.4 è stata suggerita come patologia di LQTS nei pazienti.
LQTS acquisito
Oltre al LQTS congenito, esiste anche un’altra variante di LQTS nota come ALQTS, che è causata da fattori e sostanze che riducono il flusso di potassio e compromettono la capacità del miocardio di ripolarizzare. Le condizioni ben riconosciute sono il genere femminile, l’ipopotassiemia e i farmaci che inibiscono i canali cardiaci del potassio (78-80). Un certo numero di farmaci comunemente prescritti potrebbe anche legare e bloccare preferenzialmente il canale HERG (Kv11.1, una proteina codificata dal gene KCNH2) e predispone ad ALQTS (79, 80). Recentemente, è stato dimostrato che il blocco di IKS potrebbe anche contribuire agli ALQT indotti da farmaci, specialmente quando la riserva di ripolarizzazione è compromessa (81). Fluoxetina e norfluoxetina sono stati trovati per sopprimere le proprietà IKs, sia in vivo che in vitro e ha portato a LQTS marcato (81). Polimorfismi, D85N in minK (gene KCNE1), T8A, Q9E in MiRP1 (gene KCNE2), che sono putative β-sub-unità dei canali IKs e IKr, sono stati segnalati per causare aLQTS (82, 83). LQTS autoimmune è stata riportata anche in un paziente con IgG contenenti anticorpi anti-HERG (84).
Caratteristiche elettrocardiografiche nelle tre forme comuni di sindrome del QT lungo
I tipici pattern di onde ST-T sono presenti nella maggior parte dei pazienti con LQTS genotipizzati e possono essere utilizzati per identificare i genotipi LQTS1, LQTS2 e possibilmente LQTS3 (85, 86).
La forma LQTS1 del LQTS è associata a un’ampia onda T senza accorciare l’intervallo QT all’esercizio (Figura 2A). LQTS2 è associato a onde T a bassa ampiezza, spesso bifide (Figura 2B). LQTS3 è associato a un lungo segmento iso-elettrico e a un’onda T alta a base stretta (Figura 2C). La dipendenza da pausa dell’insorgenza di TdP nelle LQTS congenite è specifica per il genotipo, essendo predominante in LQTS2 ma quasi assente in LQTS1 (87).

Figura 2. Registrazioni dell’elettrocardiogramma da pazienti con LQTS1, LQTS2 e LQTS3. (A) ECG a 12 derivazioni di un maschio di 18 anni con mutazione KCNQ1. L’intervallo QT è prolungato (QTc = ±500 ms). Il segmento ST ha una base ampia e un’ampiezza relativamente grande. L’intervallo di conduzione è normale (calibrazione standard). (B) ECG a 12 derivazioni di una ragazza di 14 anni con mutazione KCNH2. L’intervallo QT è prolungato (QTc ± 520 ms). Il segmento ST è dentellato in piombo V3 e ha un’ampiezza relativamente bassa nei conduttori delle estremità. L’intervallo di conduzione è normale (calibrazione standard). (C) ECG a 12 derivazioni di un ragazzo di 12 anni con mutazione SCN5A. L’intervallo QT è prolungato (QTc ± 600 ms). Il segmento ST ha un segmento lungo (quasi) iso-elettrico con un’onda T grande, nitida e stretta. L’intervallo di conduzione è normale (calibrazione standard).
Sebbene i modelli possano suggerire un genotipo specifico del LQTS, sono state trovate frequentemente eccezioni.
Genotipo-fenotipo
La sindrome del QT lungo è una malattia autosomica dominante. L’analisi del genotipo e del fenotipo tra i portatori di mutazione eterozigoti è stata condotta abbastanza estesamente nei pazienti LQTS1, LQTS2 e LQTS3. Durante l’infanzia, il rischio di eventi cardiaci è significativamente più alto nei maschi LQTS1 rispetto alle femmine LQTS1, mentre non sono state osservate differenze significative di genere nel rischio di eventi cardiaci tra i pazienti LQTS2 e LQTS3 (88-91). Durante l’età adulta (anche dopo i 40 anni), le femmine LQTS1 e LQTS2 potrebbero avere un rischio significativamente più elevato di eventi cardiaci rispetto ai rispettivi maschi (88-91). In generale, la letalità degli eventi cardiaci sembra essere predominante nei pazienti con LQTS3 rispetto ai pazienti con LQTS1 e LQTS2 (91). Le donne con LQTS hanno un rischio ridotto di eventi cardiaci durante la gravidanza, ma il rischio aumenta abbastanza durante il periodo postpartum di 9 mesi, specialmente nelle donne con mutazione nel gene KCNH2 (92).
La morte cardiaca improvvisa nei bambini potrebbe anche essere causata da mutazioni nei geni del canale ionico cardiaco (93-96). Circa il 28% dei bambini con una SCD inspiegabile (tra 1 e 18 anni, età media: 12,3 ± 3,8 anni) erano portatori di mutazioni nei geni causali LQTS (97). Nella SIDS, le mutazioni in SCN5A sembravano predominanti (98, 99), ma sono state trovate anche mutazioni in KCNQ1, KCNH2, KCNE2 e CAV3, SCN4B e SCN3B (100, 101). Sono state segnalate anche morti fetali intrauterine a causa di difetti nei geni del canale ionico cardiaco (12, 102).
Gestione clinica di LQTS
La cessazione di tutti i farmaci che sono noti per prolungare l’intervallo QT e anche la correzione degli squilibri elettrolitici e/o precipitare le condizioni metaboliche dovrebbe essere l’obiettivo primario durante il trattamento di pazienti LQTS (acquisiti). I sintomi in LQTS sono spesso mediati adrenergicamente, perciò la restrizione della partecipazione dei pazienti ad attività atletiche è generalmente raccomandata (24, 25). Il pilastro della terapia clinica per il LQTS è il β-blocco. Di solito vengono utilizzati preparati a lunga durata d’azione di propranololo, nadololo e metoprololo e la loro efficacia nel β-blocco viene valutata attenuando la frequenza cardiaca dell’esercizio (ad es. >20%) (24, 25). Tra tutti i β-bloccanti, propranololo e nadololo sono considerati superiori al metoprololo nei pazienti sintomatici (103). Inoltre, il β-blocco può anche essere usato come trattamento profilattico nei portatori di mutazioni silenziose per ridurre la SCD (24). In uno studio di Barsheshet et al. (30), i pazienti con mutazioni missense c-loop nel gene KCNQ1 hanno mostrato un alto rischio di eventi cardiaci potenzialmente letali e hanno avuto benefici significativi dal trattamento con β-bloccanti. Poiché le donne con LQTS2 hanno un rischio aumentato durante il periodo postpartum di 9 mesi, i β-bloccanti devono essere prescritti per ridurre qualsiasi evento cardiaco durante questo periodo ad alto rischio (92). Un defibrillatore cardioverter impiantabile (ICD) può essere preso in considerazione per i pazienti con sincope ricorrente nonostante la terapia con β-bloccante o in pazienti ad alto rischio di arresto cardiaco (ad es., LQTS2 sintomatico e LQTS3 con un prolungamento QTc documentato). La denervazione simpatica cardiaca sinistra (LCSD) è raccomandata per i pazienti con LQTS ad alto rischio in cui un ICD è controindicato o rifiutato e i β-bloccanti non sono efficaci, non tollerati, non accettati o controindicati (18, 104). I pazienti con JLNS di solito hanno un QTc > 500 ms e sono anche ad alto rischio, i β-bloccanti hanno un’efficacia insufficiente in tali pazienti nei quali è stata raccomandata una terapia precoce con ICD (18, 31).
LQT: Prospettiva saudita
Il primo rapporto sui LQTS dall’Arabia Saudita è stato pubblicato nel 1993 dall’Ospedale delle forze armate di Riyadh (105). Quattro neonati e bambini piccoli tra 6 e 48 mesi con storia di crisi ricorrenti, provenienti da un’unica famiglia, sono stati diagnosticati come LQTS (105). La storia familiare ha rivelato che altri due membri della famiglia allargata hanno avuto episodi simili di improvvisa perdita di coscienza e tre membri della famiglia sono morti improvvisamente (105). In tutti i casi, la diagnosi iniziale era l’epilessia (105). Diversi anni dopo, sono stati riportati due casi sporadici con variante relativamente più severa di LQTS neonatale combinata con blocco AV 2:1 (106, 107). Tutti questi rapporti clinici pubblicati erano senza alcun risultato genetico che potrebbe spiegare la fisiopatologia di LQTS in loro (105-107).
Noi, per la prima volta, abbiamo segnalato difetti genetici come base patologica di LQTS in pazienti simili provenienti dall’Arabia Saudita. Abbiamo studiato sei famiglie saudite con storia di sincope e morti improvvise inspiegabili di feto, neonati e bambini (12-14). LQTS1 autosomico recessivo è stato diagnosticato in bambini di due famiglie (Figura 3). LQTS2 autosomico recessivo è stato diagnosticato in due famiglie (Figura 4). In una famiglia, a una paziente di sesso femminile è stata diagnosticata LQTS2 autosomica dominante (Figura 5), il paziente ha avuto attacchi sincopali durante il periodo di recupero postpartum presso l’ospedale, che è molto comune nelle donne portatrici di mutazioni KCNH2. In tutti i nostri pazienti, l’identificazione di mutazioni patogene nei geni del canale ionico cardiaco causale LQTS ha portato a una diagnosi clinica confermata, che è stata erroneamente diagnosticata come convulsioni epilettiche prima di essere riferita a noi (13, 14) Recentemente, Shinwari et al. (15) dal King Faisal Specialist Hospital and Research Centre ha riportato una mutazione causale KCNQ1 LQTS1, H258P, in una famiglia numerosa con 12 individui affetti. Solo due portatori erano sintomatici, il loro QTc era > 500 ms e i β-bloccanti sopprimevano i sintomi clinici in un paziente e il secondo paziente sintomatico richiedeva un ICD.
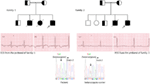
Figura 3. Disegno genealogico della famiglia-1 e 2 con LQTS1 autosomico recessivo, i probandi sono mostrati con una freccia. Gli ECG dei probandi in due famiglie sono mostrati nel mezzo, con un QTc di 557 e 529 ms, rispettivamente. Mutazione intronica, c. 387 -5 T > A nel gene KCNQ1 è stata trovata nei pazienti (mostrato in basso). La mutazione è stata mostrata con una freccia. I cerchi e i quadrati non riempiti non sono portatori della mutazione. Gli individui affetti sono mostrati come cerchi pieni (femmina) e quadrati (maschio). I quadrati e i cerchi pieni a metà sono individui con mutazione eterozigote. Gli individui deceduti sono indicati da barre, i probandi sono indicati da una freccia e il matrimonio consanguineo è indicato da = Esone − il confine dell’introne è mostrato da una linea tratteggiata con una freccia rivolta verso l’esone.
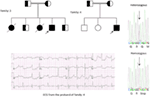
Figura 4. Disegno genealogico della famiglia-3 e 4 con LQTS2 autosomico recessivo. L’ECG del probando della famiglia-4 è mostrato sotto il disegno del pedigree, che mostra tachicardia sinusale, blocco AV quasi completo, ampio ritmo di fuga complesso con intervalli QTc molto lunghi (QT >600 ms). Mutazione, c. 3208 C > T (p. Q1070X) nel gene KCNH2 è stata trovata nei pazienti (mostrati a destra), contrassegnati da una freccia.
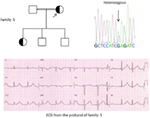
Figura 5. In alto a sinistra: pedigree della famiglia-5. L’individuo affetto è mostrato dal cerchio pieno (femmina) Il probando è indicato da una freccia. 12-condurre ECG del probando (I: 2). L’ECG mostra un alto picco, più ampio, di grande ampiezza dell’onda T, l’intervallo QTc è di 580 ms. Screening del gene KCNH2 mostra sostituzione di un nucleotide “G” per “A” (c.2362G > A, freccia), che porta alla sostituzione dell’amminoacido, p.E788K.
Genetica e dei risultati clinici nel nostro studio (12-14), Arabia Saudita, sono abbastanza intrigante per diversi motivi: (1) In totale, abbiamo studiato sei famiglie, tra i quali quattro erano omozigoti/eterozigoti composti per le mutazioni e le mutazioni nato da un’origine ancestrale; (2) Tutte le mutazioni in LQTS erano nuove, riportate solo in queste famiglie arabe; (3) A causa dell’omozigosità o eterozigosità composta per le mutazioni, i fenotipi clinici erano anche gravi nelle nostre famiglie studiate (12-14). Abbiamo suggerito che le osservazioni genetiche e fenotipiche derivassero dall’estremo alto tasso di matrimoni consanguinei in Arabia Saudita (108, 109). Il nostro studio ha fornito le prime prove scientifiche sul ruolo della consanguineità nell’esercitare un ruolo fondamentale in aritmie cardiache inspiegabili e SCDs nei bambini e negli adolescenti in Arabia Saudita (12-14). Abbiamo anche dimostrato che la mutazione KCNQ1 c.387 -5 T > A (NM_000218) (Figura 3) si era diffusa nella provincia di Assir in Arabia Saudita da un antenato comune durante diverse generazioni a causa dell’alta incidenza di matrimoni consanguinei . Finora, questa è la mutazione causale LQTS1 più comune nella popolazione saudita, che è stata osservata anche presso l’ospedale specialistico King Faisal e l’Ospedale militare Khamis Mashayt (inedito). Un risultato simile è stato ottenuto anche per la mutazione causale LQTS2, c. 3208 C > T (p.Q1070X) (Figura 4) nel gene KCNH2 (12, 14), che è anche una mutazione fondatore in Arabia Saudita e segregato durante molte generazioni nella regione Assir (vedi la mappa, Figura 6). Prevederemmo che esista un numero considerevole di individui con le mutazioni ancestrali/fondatrici menzionate (sia in KCNQ1 che KCNH2 e altri geni) in questa regione e anche nelle grandi città come Riyadh, Jeddah e Dammam a causa della migrazione urbana. Più fondatore o mutazioni ancestrali patogeni per LQTS sono molto previste in altre province dell ” Arabia Saudita a causa di alto tasso di matrimoni consanguinei.
Figura 6. Mappa dell’Arabia Saudita (per gentile concessione: Wikipedia). La regione di Assir è contrassegnata da una linea spessa, dove abbiamo trovato le mutazioni del fondatore nei geni KCNQ1 e KCNH2, descritti nella famiglia 1-4.
Poiché la LQTS è una malattia autosomica dominante, ci aspettavamo di osservare pazienti portatori di mutazioni prevalentemente eterozigoti. Ma nel nostro studio, abbiamo identificato principalmente pazienti con mutazioni recessive (12-14). Chiaramente, questi pazienti diventano prima sintomatici e, sono stati prevalentemente di cui al Principe Sultan Cardiac Centre, portandoli sotto la nostra attenzione. Tuttavia, i risultati delle nostre indagini implicano il fatto che le LQT recessive potrebbero avere fenotipi clinici fatali nei bambini, e non sono rari in Arabia Saudita (12-14). Poiché i portatori di mutazione eterozigoti sono anche suscettibili di sviluppare aritmia e le sue complicanze, dovrebbe essere presa un’iniziativa concertata per portare i medici generali locali, i cardiologi e i genetisti clinici in una piattaforma comune per identificare gli individui a rischio. Inoltre, al momento, non abbiamo anche informazioni genetiche per altri disturbi dell’aritmia familiare, ad esempio CPVT, SQTS, BrS, AF, ecc. Centri cardiogenetici specializzati dovrebbero prendere l’iniziativa di cercare i difetti genetici, mutazioni, ed eseguire studi genotipo-fenotipo in tutte le forme di aritmie ereditarie. Va anche tenuto in considerazione che non tutte le aritmie genetiche avrebbero una storia familiare, poiché in molti casi la mutazione causale dell’aritmia è di origine de novo, cioè il probando è il primo paziente in quella famiglia con la mutazione e lui/lei è la fonte per trasmettere la mutazione nelle generazioni a valle (110, 111). A causa del tasso molto elevato di matrimoni consanguinei in Arabia Saudita, ci aspettiamo molte più mutazioni fondatore esercitano un ruolo cruciale in aritmie congenite in questo paese. Identificare queste mutazioni fondatrici dovrebbe essere il nostro primo e più importante compito, che ci faciliterebbe nello sviluppo di un’efficace consulenza genetica prematrimoniale e anche pre-sintomatica in questo paese. Mutazioni nei geni causali LQTS potrebbero conferire variabilità nella penetranza clinica, in un estremo, alcuni portatori potrebbero essere presumibilmente completamente sani, ma alcuni portatori potrebbero avere la loro prima manifestazione della malattia come sincope o morte improvvisa. La morte improvvisa di un bambino o di un adulto pone una grande quantità di carico psicologico ed emotivo sulla famiglia, lo screening dei portatori è essenziale in quanto esistono farmaci semplici, ad esempio β-bloccanti (anche modificazione del comportamento), che potrebbero prevenire in modo molto efficace i portatori dalle conseguenze fatali di aritmia e SCDS. Gli individui con mutazioni omozigoti nei geni causali LQTS potrebbero avere una grave morbilità e un tasso di mortalità estremamente elevato, comprese le tachiaritmie brady-fetali e in molti casi aborti spontanei, aborti spontanei nelle madri incinte (12-14).
Non sono state condotte molte ricerche in Arabia Saudita sulla prevalenza di vari SNP nei geni legati all’aritmia e anche nei geni che li regolano. Il suo nome deriva da (112) ha descritto una variante comune S1103Y nel gene SCN5A associato all’aritmia negli afroamericani. L’allele variante chiamato Y1103 è responsabile dell’accelerazione dell’attivazione del canale, aumentando così la probabilità di aritmie cardiache in persone di origine africana (52, 112). K393N è una variante del gene KCNQ1 riportata nei pazienti LQTS1 negli Stati Uniti, ma nella popolazione araba abbiamo rilevato questa variante nel 2% degli individui (dati non pubblicati). Se la variante K393N nel gene KCNQ1 negli arabi sia paragonabile alla variante S1103Y (SCN5A) negli afro-americani o alla variante D85N (KCNE1) nella popolazione giapponese potrebbe anche meritare indagini (59).
Dichiarazione sul conflitto di interessi
Gli autori dichiarano che la ricerca è stata condotta in assenza di relazioni commerciali o finanziarie che potrebbero essere interpretate come un potenziale conflitto di interessi.
Ringraziamenti
La nostra sincera gratitudine al Prof. Arthur Wilde, Prof. Connie Bezzina, e l’editore della rivista “Cuore” per il loro permesso di utilizzare la Figura 1 in questo manoscritto da Ref. (113).
1. Bhuiyan ZA. Spettro clinico e genetico delle sindromi ereditarie di aritmia cardiaca. Amsterdam: Università di Amsterdam (2009).
2. Priori SG, Aliot E, Blømstrom-Lundqvist C, Bossaert L, Breithardt G, Brugada P, et al. Task force sulla morte cardiaca improvvisa, Società europea di cardiologia. Europace (2002) 4:3-18. doi: 10.1053 / eupc.2001.0214
CrossRef Testo integrale
3. Il sito utilizza cookie tecnici e di terze parti. Mutazioni di SCN5A associate ad un’aritmia cardiaca ereditaria, sindrome del QT lungo. Cella (1995) 80:805-11. doi:10.1016/0092-8674(95)90359-3
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
4. Curran ME, Splawski I, Timothy KW, Vincent GM, Verde ED, Keating MT. Una base molecolare per l’aritmia cardiaca: le mutazioni di HERG causano la sindrome del QT lungo. Cella (1995) 80:795-803. doi: 10.1016/0092-8674(95)90358-5
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
5. Il nostro sito utilizza cookie tecnici e di terze parti. Clonazione posizionale di un nuovo gene del canale del potassio: le mutazioni KVLQT1 causano aritmie cardiache. Nat Genet (1996) 12:17-23. doi: 10.1038 / ng0196-17
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
6. Splawski I, Tristani-Firouzi M, Lehmann MH, Sanguinetti MC, Keating MT. Le mutazioni nel gene hminK causano la sindrome del QT lungo e sopprimono la funzione IKs. Nat Genet (1997) 17:338-40. doi: 10.1038 / ng1197-338
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
7. Sanguinetti MC, Jiang C, Curran ME, Keating MT. Un legame meccanicistico tra un’aritmia cardiaca ereditata e un’aritmia cardiaca acquisita: HERG codifica il canale del potassio IKr. Cella (1995) 81:299-307. doi:10.1016/0092-8674(95)90340-2
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
8. Neyroud N, Tesson F, Denjoy I, Leibovici M, Donger C, Barhanin J, et al. Una nuova mutazione nel gene del canale del potassio KVLQT1 causa la sindrome cardioauditoria di Jervell e Lange-Nielsen. Nat Genet (1997) 15:186-9. doi: 10.1038 / ng0297-186
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
9. Kucera JP, Rohr S, Rudy Y. La localizzazione dei canali del sodio nei dischi intercalati modula la conduzione cardiaca. Circ Res (2002) 91:1176-82. doi: 10.1161 / 01.RES. 0000046237. 54156. 0 A
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
10. Oxford EM, Musa H, Maass K, Coombs W, Taffet SM, Delmar M. Connexin43 rimodellamento causato dall’inibizione dell’espressione di plakophilin-2 nelle cellule cardiache. Circ Res (2007) 101:703-11. doi:10.1161 / CIRCRESAHA.107.154252
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
11. Rohr S. Crosstalk molecolare tra giunzioni meccaniche ed elettriche al disco intercalato. Circ Res (2007) 101:637-9. doi:10.1161 / CIRCRESAHA.107.161901
Testo integrale CrossRef
12. Bhuiyan ZA, Momenah TS, Gong Q, Amin AS, Ghamdi SA, Carvalho JS, et al. Perdita fetale intrauterina ricorrente dovuta alla quasi assenza di HERG: caratterizzazione clinica e funzionale di una mutazione HERG Q1070X senza senso omozigote. Ritmo cardiaco (2008) 5:553-61. doi: 10.1016 / j. hrthm.2008.01.020
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
13. Bhuiyan ZA, Momenah TS, Amin TS, Al-Khadra AS, Alders M, Wilde AAM, et al. Una mutazione intronica che porta a un salto incompleto dell’esone-2 in KCNQ1 salva l’udito nella sindrome di Jervell e Lange-Nielsen. Prog Biophys Mol Biol (2008) 98:319-27. doi: 10.1016 / j.pbiomolbio.2008.10.004
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
14. Bhuiyan ZA, Al-Shahrani S, Al-Khadra AS, Al-Ghamdi S, Al-Kalaf K, Mannens MMAM, et al. Analisi clinica e genetica della sindrome del QT lungo nei bambini di sei famiglie in Arabia Saudita: sono diversi? Pediatr Cardiol (2009) 30:490-501. doi:10.1007 / s00246-008-9377-y
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
15. Il suo nome deriva da quello di “Al-Hazzani A”, “Dzimiri N”, “Tulbah S”, “Mallawi Y”, “Al-Fayyadh M”, et al. Identificazione di una nuova mutazione KCNQ1 in una grande famiglia saudita con sindrome del QT lungo: conseguenze cliniche e implicazioni preventive. Clin Genet (2013) 83:370-4. doi:10.1111 / j. 1399-0004. 2012.01914.x
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
16. Schwartz PJ, Stramba-Badiale M, Crotti L, Pedrazzini M, Besana A, Bosi G, et al. Prevalenza della sindrome congenita del QT lungo. Circolazione (2009) 120:1761-7. doi:10.1161 / CIRCULATIONAHA.109.863209
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
17. Per maggiori informazioni clicca qui. Criteri diagnostici per la sindrome del QT lungo. Aggiornamento. Circolazione (1993) 88:782-4. doi: 10.1161 / 01.CIR.88.2.782
Testo integrale CrossRef
18. Priori SG, Wilde AA, Horie M, Cho Y, Behr ER, Berul C, et al. Executive summary: Dichiarazione di consenso HRS / EHRA / APHRS sulla diagnosi e la gestione dei pazienti con sindromi ereditarie aritmie primarie. Europace (2013) 15:1389-406. doi: 10.1093 / europace / eut272
CrossRef Testo integrale
19. I nostri servizi sono a vostra disposizione per ogni esigenza. Registro della sindrome. Fattori di rischio per sincope ricorrente e successivi eventi fatali o quasi fatali in bambini e adolescenti con sindrome del QT lungo. J Am Coll Cardiol (2011) 57:941-50. doi: 10.1016 / j.jacc.2010.10.025
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
20. Il suo nome deriva dal greco antico, che significa “terra”, “terra”, “terra”, “terra”, “terra”, “terra”, “terra”, “terra”, “terra”, “terra”, “terra”. Spettro di mutazioni nei geni della sindrome del QT lungo. KVLQT1, HERG, SCN5A, KCNE1 e KCNE2. Circolazione (2000) 102:1178-85. doi: 10.1161 / 01.CIR.102.10.1178
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
21. Westenskow P, Splawski I, Timothy KW, Keating MT, Sanguinetti MC. Mutazioni composte: una causa comune di sindrome del QT lungo grave. Circolazione (2004) 109:1834-41. doi: 10.1161 / 01.CIR.0000125524.34234.13
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
22. Il sito utilizza cookie tecnici e di terze parti per migliorare la tua esperienza di navigazione. Rischio di eventi cardiaci potenzialmente letali nei pazienti con sindrome del QT lungo e mutazioni multiple. Ritmo cardiaco (2013) 10:378-82. doi: 10.1016 / j. hrthm.2012.11.006
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
23. Napolitano C, Priori SG, Schwartz PJ, Bloise R, Ronchetti E, Nastoli J, et al. Test genetici nella sindrome del QT lungo: sviluppo e validazione di un approccio efficiente alla genotipizzazione nella pratica clinica. JAMA (2005) 294:2975-80. doi:10.1001 / jama.294.23.2975
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
24. Roden DM. Pratica clinica. Sindrome del QT lungo. N Engl J Med (2008) 358:169-76. doi:10.1056 / NEJMcp0706513
CrossRef Testo completo
25. Il nostro sito utilizza cookie tecnici e di terze parti per migliorare la tua esperienza di navigazione. Correlazione genotipo-fenotipo nella sindrome del QT lungo: trigger gene-specifici per aritmie potenzialmente letali. Circolazione (2001) 103:89-95. doi: 10.1161 / 01.CIR.103.1.89
Testo integrale CrossRef
26. Ackerman MJ, Tester DJ, Porter CJ. Nuoto, un trigger aritmogenico gene-specifico per la sindrome del QT lungo ereditata. Mayo Clin Proc (1999) 74:1088-94. doi:10.4065/74.11.1088
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
27. Kapa S, Tester DJ, Salisbury BA, Harris-Kerr C, Pungliya MS, Alders M, et al. Test genetici per la sindrome del QT lungo: distinguere mutazioni patogene da varianti benigne. Circolazione (2009) 120:1752-60. doi:10.1161 / CIRCULATIONAHA.109.863076
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
28. Moss AJ, Shimizu W, Wilde AA, Towbin JA, Zareba W, Robinson JL, et al. Aspetti clinici della sindrome del QT lungo di tipo 1 per posizione, tipo codificante e funzione biofisica delle mutazioni che coinvolgono il gene KCNQ1. Circolazione (2007) 115:2481-9. doi:10.1161 / CIRCULATIONAHA.106.665406
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
29. Amin AS, Giudicessi JR, Tijsen AJ, Spanjaart AM, Reckman YJ, Klemens CA, et al. Varianti nella regione 3 ‘ non tradotta del canale del potassio Kv7.1 codificato KCNQ1 modificano la gravità della malattia nei pazienti con sindrome del QT lungo di tipo 1 in modo allele-specifico. Eur Cuore J (2012) 33:714-23. doi: 10.1093/eurheartj / ehr473
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
30. Barsheshet A, Goldenberg I, O-Uchi J, Moss AJ, Jons C, Shimizu W, et al. Mutazioni nelle anse citoplasmatiche del canale KCNQ1 e rischio di eventi potenzialmente letali: implicazioni per la risposta specifica alla mutazione alla terapia β-bloccante nella sindrome del QT lungo di tipo 1. Circolazione (2012) 125:1988-96. doi:10.1161 / CIRCULATIONAHA.111.048041
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
31. Schwartz PJ, Spazzolini C, Crotti L, Bathen J, Amlie JP, Timothy K, et al. La sindrome di Jervell e Lange-Nielsen: storia naturale, base molecolare ed esito clinico. Circolazione (2006) 113:783-90. doi:10.1161 / CIRCULATIONAHA.105.592899
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
32. Bhuiyan ZA, Wilde AA. IKs nel cuore e nell’udito, l’orecchio può fare con meno del cuore. Circ Cardiovasc Genet (2013) 6:141-3. doi:10.1161 / CIRCGENETICS.113.000143
Testo integrale CrossRef
33. Wollnik B, Schroeder BC, Kubisch C, Esperer HD, Wieacker P, Jentsch TJ. Meccanismi fisiopatologici delle mutazioni dominanti e recessive del canale KVLQT1 K + riscontrate nelle aritmie cardiache ereditarie. Hum Mol Genet (1997) 6: 1943-9. doi: 10.1093 / hmg / 6.11.1943
CrossRef Testo integrale
34. Wilde AA, Jongbloed RJ, Doevendans PA, Düren DR, Hauer RN, van Langen IM, et al. Gli stimoli uditivi come trigger per eventi aritmici differenziano i pazienti correlati a HERG (LQTS2) dai pazienti correlati a KVLQT1 (LQTS1). J Am Coll Cardiol (1999) 33:327-32. doi:10.1016 / S0735-1097(98)00578-6
Testo integrale CrossRef
35. Il nostro sito utilizza cookie tecnici e di terze parti. Aumento del rischio di eventi aritmici nella sindrome del QT lungo con mutazioni nella regione dei pori del canale del potassio del gene umano etere-a-go-go-correlato. Circolazione (2002) 105:794-9. doi: 10.1161 / hc0702.105124
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
36. Lupoglazoff JM, Denjoy I, Villain E, Fressart V, Simon F, Bozio A, et al. Sindrome del QT lungo nei neonati: disturbi della conduzione associati a mutazioni HERG e bradicardia sinusale con mutazioni KCNQ1. J Am Coll Cardiol (2004) 43:826-30. doi: 10.1016 / j.jacc.2003.09.049
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
37. Il nostro sito utilizza cookie tecnici e di terze parti. Torsades de pointes che complicano il blocco atrioventricolare: evidenza di una predisposizione genetica. Ritmo cardiaco (2007) 4:170-4. doi: 10.1016 / j. hrthm.2006.10.004
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
38. Hoorntje T, Alders M, van Tintelen P, van der Lip K, Sreeram N, van der Wal A, et al. Omozigote troncamento prematuro della proteina HERG: l’HERG knockout umano. Circolazione (1999) 100:1264-7. doi: 10.1161 / 01.CIR.100.12.1264
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
39. Piippo K, Laitinen P, Swan H, Toivonen L, Viitasalo M, Pasternack M, et al. L’omozigosi per una mutazione del canale del potassio HERG causa una forma grave di sindrome del QT lungo: identificazione di un’apparente mutazione del fondatore nei finlandesi. J Am Coll Cardiol (2000) 35:1919-25. doi:10.1016 / S0735-1097(00)00636-7
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
40. I nostri servizi sono a vostra disposizione per ogni esigenza. Caratterizzazione clinica, genetica e biofisica di una mutazione HERG omozigote che causa una grave sindrome del QT lungo neonatale. Pediatr Res (2003) 53:744-8. doi: 10.1203 / 01.PDR.0000059750.17002.B6
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
41. Ackerman MJ, Tester DJ, Jones GS, Will ML, Burrow CR, Curran ME. Differenze etniche nelle varianti del canale cardiaco del potassio: implicazioni per la suscettibilità genetica alla morte cardiaca improvvisa e test genetici per la sindrome congenita del QT lungo. Mayo Clin Proc (2003) 78:1479-87. doi:10.4065/78.12.1479
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
42. Crotti L, Lundquist AL, Insolia R, Pedrazzini M, Ferrandi C, De Ferrari GM, et al. KCNH2-K897T è un modificatore genetico della sindrome congenita latente del QT lungo. Circolazione (2005) 112:1251-8. doi:10.1161 / CIRCULATIONAHA.105.549071
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
43. Nof E, Cordeiro JM, Pérez GJ, Scornik FS, Calloe K, Love B, et al. Un polimorfismo a singolo nucleotide comune può esacerbare la sindrome del QT lungo di tipo 2 che porta alla morte improvvisa del neonato. Circ Cardiovasc Genet (2010) 3:199-206. doi:10.1161 / CIRCGENETICS.109.898569
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
44. Bezzina C, Veldkamp MW, van Den Berg MP, Postma AV, Rook MB, Viersma JW, et al. Una singola mutazione del canale Na(+) che causa sia le sindromi long-QT che Brugada. Circ Res (1999) 85:1206-13. doi: 10.1161 / 01.RES.85.12.1206
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
45. Kyndt F, Probst V, Potet F, Demolombe S, Chevallier JC, Baro I, et al. Nuova mutazione SCN5A che porta a un difetto di conduzione cardiaca isolato o alla sindrome di Brugada in una grande famiglia francese. Circolazione (2001) 104:3081-6. doi: 10.1161 / hc5001.100834
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
46. Makita N, Behr E, Shimizu W, Horie M, Sunami A, Crotti L, et al. La mutazione E1784K in SCN5A è associata al fenotipo clinico misto della sindrome del QT lungo di tipo 3. J Clin Investire (2008) 118:2219-29. doi: 10.1172 / JCI34057
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
47. Keller DI, Acharfi S, Delacrétaz E, Benammar N, Rotter M, Pfammatter JP, et al. Una nuova mutazione in SCN5A, delQKP 1507-1509, che causa la sindrome del QT lungo: ruolo del residuo Q1507 nell’inattivazione del canale del sodio. J Mol Cell Cardiol (2003) 35:1513-21. doi: 10.1016 / jjmcc.2003.08.007
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
48. Priori SG, Schwartz PJ, Napolitano C, Bloise R, Ronchetti E, Grillo M, et al. Stratificazione del rischio nella sindrome del QT lungo. N Engl J Med (2003) 348:1866-74. doi:10.1056 / NEJMoa022147
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
49. Lupoglazoff JM, Cheav T, Baroudi G, Berthet M, Denjoy I, Cauchemez B, et al. Mutazione omozigote di SCN5A nella sindrome del QT lungo con blocco atrioventricolare funzionale da due a uno. Circ Res (2001) 89:E16–21. doi: 10.1161 / hh1401.095087
CrossRef Testo integrale
50. Shi R, Zhang Y, Yang C, Huang C, Zhou X, Qiang H, et al. La mutazione cardiaca del canale del sodio delQKP 1507-1509 è associata allo spettro fenotipico in espansione di LQT3, disturbo della conduzione, cardiomiopatia dilatativa e alta incidenza di morte improvvisa giovanile. Europace (2008) 10:1329-35. doi: 10.1093/europace / eun202
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
51. Zumhagen S, Veldkamp MW, Stallmeyer B, Baartscheer A, Eckardt L, Paul M, et al. Una mutazione eterozigote della delezione nel gene cardiaco del canale del sodio SCN5A con caratteristiche di perdita e guadagno di funzione si manifesta come malattia di conduzione isolata, senza segni di Brugada o sindrome del QT lungo. PLoS Uno (2013) 8:e67963. doi:10.1371 / giornale.pone.0067963
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
52. Il nostro sito utilizza cookie tecnici e di terze parti. Una variante comune del canale cardiaco del sodio associata alla morte improvvisa infantile negli afroamericani, SCN5A S1103Y. J Clin Invest (2006) 116:430-5. doi: 10.1172 / JCI25618
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
53. Il sito utilizza cookie tecnici e di terze parti per migliorare la tua esperienza di navigazione. La mutazione Ankyrin-B causa aritmia cardiaca di tipo 4 lungo QT e morte cardiaca improvvisa. Natura (2003) 421:634-9. doi: 10.1038 / nature01335
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
54. Schott JJ, Charpentier F, Peltier S, Foley P, Drouin E, Bouhour JB, et al. Mappatura di un gene per la sindrome del QT lungo al cromosoma 4q25-27. Am J Hum Genet (1995) 57:1114-22.
Pubmed Abstract / Pubmed Testo completo
55. Le proteine Barhanin J, Lesage F, Guillemare E, Fink M, Lazdunski M, Romey GK(V)LQT1 e LSK (minK) si associano per formare la corrente cardiaca di potassio I(Ks). Natura (1996) 384:78-80. doi: 10.1038 | 384078a0
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
56. Sanguinetti MC, Curran ME, Zou A, Shen J, Spector PS, Atkinson DL, et al. Assemblaggio di proteine K(V)LQT1 e minK (IsK) per formare il canale cardiaco I(Ks) del potassio. Natura (1996) 384:80-3. doi: 10.1038 / 384080a0
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
57. Schulze-Bahr E, Wang Q, Wedekind H, Haverkamp W, Chen Q, Sun Y, et al. Le mutazioni KCNE1 causano la sindrome di Jervell e Lange-Nielsen. Nat Genet (1997) 17:267-8. doi: 10.1038 / ng1197-267
CrossRef Testo completo
58. Duggal P, Vesely MR, Wattanasirichaigoon D, Villafane J, Kaushik V, Beggs AH. Mutazione del gene per IKS associato sia con Jervell e Lange-Nielsen e Romano-Ward forme di sindrome del QT lungo. Circolazione (1998) 97:142-6. doi: 10.1186/1471-2350-9-24
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
59. Nishio Y, Makiyama T, Itoh H, Sakaguchi T, Ohno S, Gong YZ, et al. D85N, un polimorfismo KCNE1, è una variante del gene che causa la malattia nella sindrome del QT lungo. J Am Coll Cardiol (2009) 54:812-9. doi: 10.1016 / j.jacc.2009.06.005
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
60. Paulussen AD, Gilissen RA, Armstrong M, Doevendans PA, Verhasselt P, Smeets HJ, et al. Variazioni genetiche di KCNQ1, KCNH2, SCN5A, KCNE1 e KCNE2 nei pazienti con sindrome del QT lungo indotta da farmaci. Mol Med (2004) 82:182-8. doi:10.1007 / s00109-003-0522-z
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
61. Abbott GW, Sesti F, Splawski I, Buck ME, Lehmann MH, Timothy KW, et al. MiRP1 forma canali di potassio IKr con HERG ed è associato ad aritmia cardiaca. Cella (1999) 97:175-87. doi: 10.1016 / S0092-8674 (00) 80728-X
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
62. Il sito utilizza cookie tecnici e di terze parti. Una mutazione KCNE2 in un paziente con aritmia cardiaca indotta da stimoli uditivi e squilibrio elettrolitico sierico. Cardiovasc Res (2008) 77:98-106. doi: 10.1093/cvr / cvm030
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
63. Gesso NM, Tawil R, Tristani-Firouzi M, Canún S, Bendahhou S, Tsunoda A, et al. Le mutazioni in Kir2.1 causano i fenotipi elettrici evolutivi ed episodici della sindrome di Andersen. Cella (2001) 105:511-9. doi: 10.1016 / S0092-8674(01)00342-7
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
64. Kubo Y, Baldwin TJ, Jan YN, Jan LY. Struttura primaria ed espressione funzionale di un canale del potassio del raddrizzatore interno del topo. Natura (1993) 362:127-33. doi: 10.1038 | 362127a0
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
65. I nostri servizi sono a vostra disposizione. Clonazione molecolare ed espressione di un canale di potassio raddrizzatore interno del cuore umano. Neuroreport (1994) 5:2501-5. doi: 10.1097/00001756-199412000-00024
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
66. Il sito utilizza cookie tecnici e di terze parti. La disfunzione del canale del calcio Ca(V)1.2 causa un disturbo multisistemico tra cui aritmia e autismo. Cella (2004) 119:19-31. doi: 10.1016 / j.cella.2004.09.011
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
67. I nostri servizi sono a vostra disposizione per ogni esigenza. Grave disturbo da aritmia causato da mutazioni cardiache dei canali del calcio di tipo L. Proc Natl Acad Sci U S A (2005) 102:8089-96. doi: 10.1073 / pnas.0502506102
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
68. Gillis J, Burashnikov E, Antzelevitch C, Blaser S, Gross G, Turner L, et al. QT lungo, sindattilia, contratture articolari, ictus e nuova mutazione CACNA1C: espansione dello spettro della sindrome di Timothy. Sono J Med Genet Un (2012) 158UN:182-7. doi:10.1002 / ajmg.a.34355
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
69. Dufendach KA, Giudicessi JR, Boczek NJ, Ackerman MJ. Il mosaicismo materno confonde la diagnosi neonatale della sindrome di Timothy di tipo 1. Pediatria (2013) 131:e1991-5. doi:10.1542 / peds.2012-2941
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
70. Il sito utilizza cookie tecnici e di terze parti. La caveolina mutante-3 induce la corrente tardiva persistente del sodio ed è associata con la sindrome lungo-QT. Circolazione (2006) 114:2104–12. doi:10.1161 / CIRCULATIONAHA.106.635268
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
71. I nostri servizi sono a vostra disposizione per ogni esigenza. Meccanismo novello per la sindrome di morte infantile improvvisa: corrente tardiva persistente del sodio secondaria alle mutazioni in caveolin-3. Ritmo cardiaco (2007) 4:161-6. doi: 10.1016 / j. hrthm.2006.11.030
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
72. Engelman JA, Zhang X, Galbiati F, Volonte D, Sotgia F, Pestell RG, et al. Genetica molecolare della famiglia genica della caveolina: implicazioni per tumori umani, diabete, malattia di Alzheimer e distrofia muscolare. Am J Hum Genet (1998) 63: 1578-87. doi:10.1086/302172
Testo integrale CrossRef
73. I nostri servizi sono a vostra disposizione. La localizzazione dei canali Ca(2+) di tipo L cardiaco in un complesso di segnalazione macromolecolare caveolare è necessaria per la regolazione beta(2)-adrenergica. Proc Natl Acad Sci U S A (2006) 103:7500-5. doi: 10.1073 / pnas.0503465103
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
74. Medeiros-Domingo A, Kaku T, Tester DJ, Iturralde-Torres P, Itty A, Ye B, et al. Subunità beta4 del canale del sodio codificata SCN4B nella sindrome congenita del QT lungo. Circolazione (2007) 116:134-42. doi:10.1161 / CIRCULATIONAHA.106.659086
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
75. Il nostro sito utilizza cookie tecnici e di terze parti. La mutazione di una proteina A-chinasi-ancoraggio causa la sindrome del QT lungo. Proc Natl Acad Sci U S A (2007) 104:20990-5. doi: 10.1073 / pnas.0710527105
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
76. Il sito utilizza cookie tecnici e di terze parti. Mutazione alfa-1-syntrophin e sindrome del QT lungo: una malattia di interruzione del canale del sodio. Circ Arrhythm Electrophysiol (2008) 1:193-201. doi:10.1161 / CIRCEP.108.769224
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
77. Yang Y, Yang Y, Liang B, Liu J, Li J, Grunnet M, et al. Identificazione di un Kir3.4 mutazione nella sindrome congenita del QT lungo. Sono J Hum Genet (2010) 86:872-80. doi:10.1016 / j.ajhg.2010.04.017
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
78. Roden DM. Meccanismi e gestione della proaritmia. Am J Cardiol (1998) 82:49I-57I. doi: 10.1016 / S0002-9149 (98)00472-X
CrossRef Testo completo
79. Mitcheson JS, Chen J, Lin M, Culberson C, Sanguinetti MC. Una base strutturale per la sindrome del QT lungo indotta da farmaci. Proc Natl Acad Sci U S A (2000) 97:12329-33. doi: 10.1073 / pnas.210244497
Testo integrale CrossRef
80. Kannankeril PJ, Roden DM. QT lungo indotto da farmaci e torsade de pointes: recenti progressi. Curr Opin Cardiol (2007) 22:39-43. doi: 10.1097 / HCO.0b013e32801129eb
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
81. Veerman CC, Verkerk AO, Blom MT, Klemens CA, Langendijk PN, van Ginneken AC, et al. Il blocco di corrente di potassio del raddrizzatore ritardato lento contribuisce in modo importante alla sindrome del QT lungo indotta da farmaci. Circ Arrhythm Electrophysiol (2013) 6:1002-9. doi:10.1161 / CIRCEP.113.000239
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
82. I nostri servizi sono a vostra disposizione per ogni esigenza. Un polimorfismo comune associato ad aritmia cardiaca indotta da antibiotici. Proc Natl Acad Sci U S A (2000) 97:10613-8. doi: 10.1073 / pnas.180223197
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
83. Kääb S, Crawford DC, Sinner MF, Behr ER, Kannankeril PJ, Wilde AA, et al. Un ampio sondaggio sui geni candidati identifica il polimorfismo KCNE1 D85N come un possibile modulatore di torsioni di punta indotte da farmaci. Circ Cardiovasc Genet (2012) 5:91-9. doi:10.1161 / CIRCGENETICS.111.960930
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
84. Il suo nome deriva dal latino “Kusano KF”, “Haraoka K”, “Tani Y”, “Nagase S”, et al. Sindrome del QT lungo indotta da anticorpi anti-KCNH2: nuova forma acquisita di sindrome del QT lungo. J Am Coll Cardiol (2007) 50:1808-9. doi: 10.1016 / j.jacc.2007.07.037
CrossRef Testo completo
85. Il nostro sito utilizza cookie tecnici e di terze parti. Modelli di onde T ECG in forme geneticamente distinte della sindrome del QT lungo ereditario. Circolazione (1995) 92:2929-34. doi: 10.1161 / 01.CIR.92.10.2929
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
86. Zhang L, Timothy KW, Vincent GM, Lehmann MH, Fox J, Giuli LC, et al. Spettro dei pattern dell’onda ST-T e parametri di ripolarizzazione nella sindrome congenita del QT lungo: i risultati dell’ECG identificano i genotipi. Circolazione (2000) 102:2849-55. doi: 10.1161 / 01.CIR.102.23.2849
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
87. Tan HL, Bardai A, Shimizu W, Moss AJ, Schulze-Bahr E, Noda T, et al. Insorgenza genotipo-specifica di aritmie nella sindrome congenita del QT lungo: possibili implicazioni terapeutiche. Circolazione (2006) 114:2096-103. doi:10.1161 / CIRCULATIONAHA.106.642694
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
88. Locati EH, Zareba W, Moss AJ, Schwartz PJ, Vincent GM, Lehmann MH, et al. Differenze legate all’età e al sesso nelle manifestazioni cliniche in pazienti con sindrome congenita del QT lungo: risultati del Registro internazionale LQTS. Circolazione (1998) 97:2237-44. doi: 10.1161 / 01.CIR.97.22.2237
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
89. Il sito utilizza cookie tecnici e di terze parti per migliorare la tua esperienza di navigazione. Influenza del genotipo sul decorso clinico della sindrome del QT lungo. International Long-QT Syndrome Registry Research Group. N Engl J Med (1998) 339:960-5. doi: 10.1056 / NEJM199810013391404
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
90. Il nostro sito utilizza cookie tecnici e di terze parti. Sindrome del QT lungo dopo i 40 anni. Circolazione (2008) 117:2192-201. doi:10.1161 / CIRCULATIONAHA.107.729368
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
91. Il nostro sito utilizza cookie tecnici e di terze parti. Registro della sindrome. Effetti modulanti di età e sesso sul decorso clinico della sindrome del QT lungo per genotipo. J Am Coll Cardiol (2003) 42:103-9. doi:10.1016 / S0735-1097(03)00554-0
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
92. Il nostro sito utilizza cookie tecnici e di terze parti. Sindrome del QT lungo e gravidanza. J Am Coll Cardiol (2007) 49:1092-8. doi: 10.1016 / j.jacc.2006.09.054
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
93. Schwartz PJ, Priori SG, Dumaine R, Napolitano C, Antzelevitch C, Stramba-Badiale M, et al. Un legame molecolare tra la sindrome della morte improvvisa infantile e la sindrome del QT lungo. N Engl J Med (2000) 343:262-7. doi:10.1056 / NEJM200007273430405
CrossRef Testo completo
94. Il suo nome deriva dal greco antico, che significa “terra”, “terra”, “terra”, “terra”, “terra”, “terra”, “terra”, “terra”, “terra”, “terra”, “terra”, “terra”. Diagnosi molecolare in un bambino con sindrome da morte improvvisa infantile. Lancet (2001) 358:1342-3. doi:10.1016 / S0140-6736(01)06450-9
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
95. Christiansen M, Tønder N, Larsen LA, Andersen PS, Simonsen H, Oyen N, et al. Mutazioni nel canale HERG K+-ion: un nuovo legame tra sindrome del QT lungo e sindrome della morte improvvisa infantile. Am J Cardiol (2005) 95:433-4. doi:10.1016 / j. amjcard.2004.09.054
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
96. Il DJ del tester, Ackerman MJ. Sindrome del QT lungo post mortem test genetici per morte improvvisa inspiegabile nei giovani. J Am Coll Cardiol (2007) 49:240-6. doi: 10.1016 / j.jacc.2006.10.010
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
97. Hofman N, Tan HL, Clur SA, Alders M, van Langen IM, Wilde AA. Contributo della malattia cardiaca ereditaria alla morte cardiaca improvvisa durante l’infanzia. Pediatria (2007) 120:e967-73. doi:10.1542 / peds.2006-3751
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
98. Il nostro obiettivo è quello di migliorare la qualità e la qualità del prodotto. Mutazione de novo nel gene SCN5A associata all’insorgenza precoce della morte improvvisa infantile. Circolazione (2001) 104:1158-64. doi: 10.1161 / hc3501.095361
CrossRef Testo completo
99. Wang DW, Desai RR, Crotti L, Arnestad M, Insolia R, Pedrazzini M, et al. Disfunzione cardiaca del canale del sodio nella sindrome da morte improvvisa infantile. Circolazione (2007) 115:368-76. doi:10.1161 / CIRCULATIONAHA.106.646513
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
100. Van Norstrand DW, Valdivia CR, Tester DJ, Ueda K, London B, Makielski JC, et al. Caratterizzazione molecolare e funzionale di nuove mutazioni del gene simile al glicerolo-3-fosfato deidrogenasi 1 (GPD1-L) nella sindrome da morte improvvisa infantile. Circolazione (2007) 116:2253-9. doi: 10.1161 / CIRCULATIONAHA.107.704627
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
101. Tan BH, Pundi KN, Van Norstrand DW, Valdivia CR, Tester DJ, Medeiros-Domingo A, et al. Mutazioni associate alla sindrome della morte improvvisa infantile nelle subunità beta del canale del sodio. Ritmo cardiaco (2010) 7:771-8. doi: 10.1016 / j. hrthm.2010.01.032
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
102. Miller TE, Estrella E, Myerburg RJ, Garcia de Viera J, Moreno N, Rusconi P, et al. Perdita fetale ricorrente del terzo trimestre e mosaicismo materno per la sindrome del QT lungo. Circolazione (2004) 109:3029-34. doi: 10.1161 / 01.CIR.0000130666.81539.9 E
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
103. Chockalingam P, Crotti L, Girardengo G, Johnson JN, Harris KM, van der Heijden JF, et al. Non tutti i beta-bloccanti sono uguali nella gestione dei tipi di sindrome del QT lungo 1 e 2: recidive più elevate di eventi sotto Metoprololo. J Am Coll Cardiol (2012) 60:2092-6. doi: 10.1016 / j.jacc.2012.07.046
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
104. Schwartz PJ, Priori SG, Cerrone M, Spazzolini C, Odero A, Napolitano C, et al. Denervazione simpatica cardiaca sinistra nella gestione di pazienti ad alto rischio affetti dalla sindrome del QT lungo. Circolazione (2004) 109:1826-33. doi: 10.1161 / 01.CIR.0000125523.14403.1 E
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
105. Singh B, al Shahwan SA, Habbab MA, al Deeb SM, Biary N. Sindrome idiopatica del QT lungo: fare la domanda giusta. Lancetta (1993) 341:741. doi:10.1016/0140-6736(93)90501-7
Testo integrale CrossRef
106. Gorgels AP, Al Fadley F, Zaman L, Kantoch MJ, Al Halees Z. La sindrome del QT lungo con compromissione della conduzione atrioventricolare: una variante maligna nei neonati. J Cardiovasc Electrophysiol (1998) 9:1225-32. doi: 10.1111 / j. 1540-8167. 1998.tb00096.x
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
107. Kantoch MJ, Qurashi MM, Bulbul ZR, Gorgels AP. Un neonato con una complessa cardiopatia congenita, blocco atrioventricolare e tachicardia ventricolare a torsione di punta. Stimolazione Clin Electrophysiol (1998) 21:2664-7. doi: 10.1111 / j. 1540-8159. 1998.tb00043.x
CrossRef Testo completo
108. Al-Hazmi MA, al-Swailem AR, Warsy AS, al-Swailem AM, Sulaimani R, al-Meshari AA. Consanguineità tra la popolazione saudita. J Med Genet (1995) 32:623-6. doi: 10.1136 / jmg.32.8.623
Testo integrale CrossRef
109. El Mouzan MI, Al Salloum AA, Al Herbish AS, Qurachi MM, Al Omar AA. Consanguineità e principali malattie genetiche nei bambini sauditi: uno studio trasversale basato sulla comunità. Ann Saudi Med (2008) 28:169-73. doi:10.4103/0256-4947.51726
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
110. Medeiros-Domingo A, Bhuiyan ZA, Tester DJ, Hofman N, Bikker H, van Tintelen JP, et al. Il recettore RYR2-codificato ryanodine / canale di rilascio del calcio in pazienti diagnosticati in precedenza sia con tachicardia ventricolare polimorfa catecolaminergica o genotipo negativo, sindrome del QT lungo indotta da esercizio: un’analisi mutazionale completa del telaio di lettura aperto. J Am Coll Cardiol (2009)54: 2065-74. doi: 10.1016 / j.jacc.2009.08.022
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
111. Al-Aama JY, Al-Ghamdi S, Bdier AY, Wilde AA, Bhuiyan ZA. Mutazione de novo nel gene KCNQ1 causale alla sindrome di Jervell e Lange-Nielsen. Clin Genet (2013). doi: 10.1111 / cge.12300
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
112. Il nostro sito utilizza cookie tecnici e di terze parti per migliorare la tua esperienza di navigazione. Variante del canale del sodio SCN5A implicato nel rischio di aritmia cardiaca. Scienza (2002) 297:1333-6. doi: 10.1126 / scienza.1073569
Testo integrale CrossRef
113. Wilde AA, Bezzina CR. Genetica delle aritmie cardiache. Cuore (2005) 91:1352-8.