Mucopolisaccaridosi: segni diagnostici precoci nei neonati e nei bambini
Le mucopolisaccaridosi (MPS) sono un gruppo di malattie clinicamente eterogenee causate da carenze degli enzimi lisosomiali necessari per la scomposizione dei glicosaminoglicani (GAG).
I MPS sono caratterizzati da manifestazioni cliniche progressive e sistemiche. Nonostante la loro eterogeneità biochimica e genetica, diversi tipi condividono caratteristiche cliniche chiave in diverse combinazioni, tra cui displasia articolare e scheletrica con rigidità (eccetto MPS IV dove c’è lassità) e dolore, caratteristiche facciali grossolane, annebbiamento corneale, ernie inguinali o addominali, infezioni ricorrenti del tratto respiratorio superiore, malattia della valvola cardiaca, sindrome del tunnel carpale e coinvolgimento neurologico variabile. Queste caratteristiche compaiono solitamente nei primi mesi di vita per le forme gravi e nella prima infanzia per le forme più attenuate, ma sono spesso sottovalutate e generalmente prese in considerazione solo quando chiaramente evidenti.
La rarità di questi disturbi e la variabilità nella presentazione clinica porta frequentemente ad un ritardo diagnostico; questo può variare da mesi, per le forme gravi, ad anni, per quelle attenuate (vedi Rigoldi et al. in questo supplemento). Tuttavia, a causa della rapida progressione e della necessità urgente di intervento nelle presentazioni gravi, anche mesi di ritardo nella diagnosi possono produrre risultati catastrofici per la salute futura del paziente.
Il nostro obiettivo è quello di sottolineare in questa recensione i primissimi segni delle forme gravi che dovrebbero allertare il pediatra alla loro prima comparsa.
Quali segni / sintomi possiamo aspettarci nei bambini con forme gravi di MPS?
I sintomi e i segni indicativi di diverse forme di MPS sono riportati nella Tabella 1 in base all’età. Nei primi 6 mesi di vita, ernie inguinali, infezioni respiratorie anormalmente frequenti, otite e organomegalia possono essere osservate nei pazienti con MPS, in particolare nella MPS I (sindrome di Hurler) che ha l’esordio più precoce e grave. Inoltre, da 6 a 12 mesi, questi bambini sviluppano molto spesso un gibbus (cifosi toraco-lombare), soffio cardiaco, ernia ombelicale, ipotonia lieve e ritardo della crescita . Possono anche avere il tipico aspetto facciale grossolano (Fig. 1). MPS II, VI e VII possono presentarsi con un fenotipo precoce simile. I pazienti con MPS IV hanno un fenotipo scheletrico precoce con comparsa di displasia dell’anca nei primi mesi di vita e petto di piccione (pectus carinatum) nei primi 12-18 mesi. I neonati MPS II presentano un coinvolgimento simile ma con esordio successivo. Nel secondo anno di vita, MPS I Hurler sindrome neonati sviluppano ritardo cognitivo, mentre un paziente MPS II potrebbe essere identificato solo a causa della crescita eccessiva, infezioni delle vie aeree frequenti, e interventi chirurgici multipli ; le caratteristiche tipiche del viso appaiono solo più tardi. Alcuni pazienti di MPS II inoltre sviluppano l’iperattività fra 18 e 24 mesi. Nel terzo anno di vita, iperattività, ritardo cognitivo, sono facilmente riconoscibili in MPS II e MPS III.
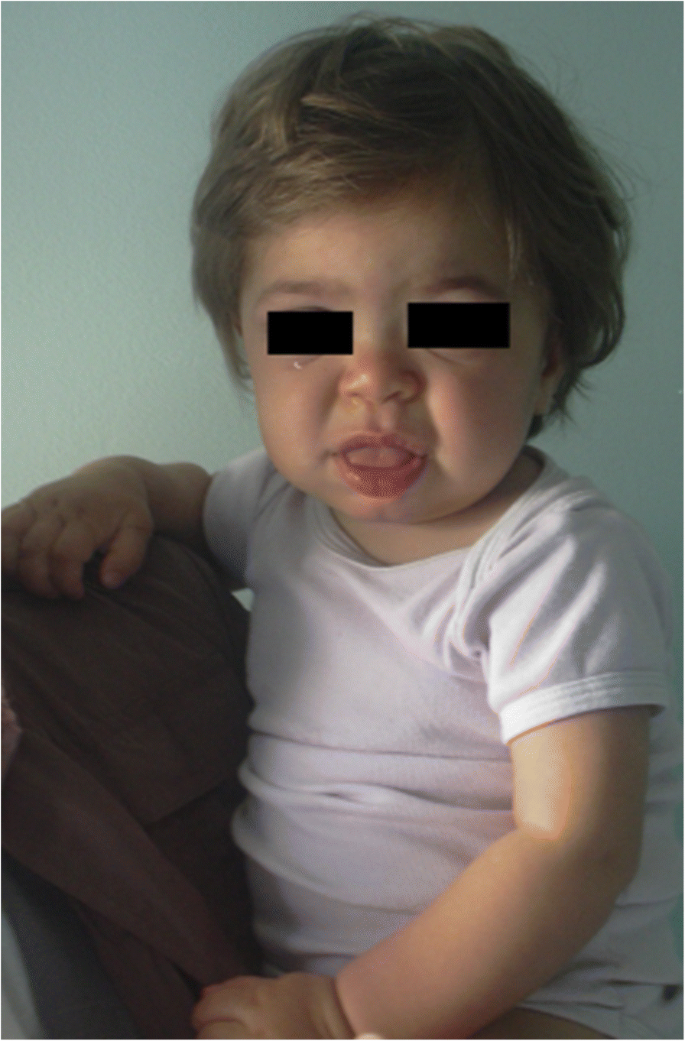
MPS IH nel paziente di 1 anno. Facies grossolana: ponte nasale piatto, macroglossia, bossing frontale
In sintesi, le manifestazioni scheletriche sono tipiche e talvolta precoci. La facies grossolana e il ritardo cognitivo non sono segni preminenti precoci.
Quali sono le diverse presentazioni delle varie tipologie di MPS?
MPS con coinvolgimento somatico e cognitivo
Mucopolisaccaridosi di tipo I (MPS I; OMIM #252800) (Figs. 1, 2, 3, 4 e 5) è causato da una carenza dell’idrolasi lisosomiale α-l-iduronidasi, che porta all’accumulo multisistemico di due GAG: dermatan solfato (DS) e eparan solfato (HS) . In base alla sua gravità, MPS I è tradizionalmente diviso in tre fenotipi clinici; tuttavia, queste forme rappresentano un continuum di gravità della malattia. La sindrome di Hurler, la forma più grave, si presenta tipicamente durante il primo anno di vita. I bambini affetti sviluppano rapidamente un significativo deterioramento cognitivo e una malattia somatica nei sistemi di organi multipli, portando alla morte entro il primo decennio in assenza di trattamento. Le forme attenuate di MPS I, note come sindrome di Hurler–Scheie e sindrome di Scheie, rispettivamente, sono caratterizzate da insorgenza successiva dei sintomi, aspettativa di vita più lunga e coinvolgimento lieve o assente del sistema nervoso centrale (SNC). L’ereditarietà è autosomica recessiva e in almeno il 50% dei casi la previsione del fenotipo è possibile sulla base del genotipo mentre nel restante 50% vengono rilevate nuove mutazioni . La prevalenza è di circa 1 su 100.000 nati vivi .

MPS IH. un Gibbus in un paziente di 9 mesi. b Radiografia della colonna vertebrale in un paziente di 3 anni. immagine di risonanza magnetica ponderata c T2 della colonna vertebrale che mostra cifosi significativa e stenosi del canale vertebrale

MPS IH. Prova a raggi X di dysostosis multiplex. a Ispessimento delle costole in un paziente di 3 anni e b displasia dell’anca in un altro paziente di età compresa tra 18 mesi

MPS IH: una mano artiglio in un paziente di 18 mesi; b mano X-ray.

MPS IH. un ispessimento dell’osso corticale del cranio e anormale conformazione “a forma di J” della sella turcica. Dilatazione degli spazi periventricolari in b FLAIR e c T-2 immagini di risonanza magnetica ponderate di un paziente di 17 mesi
Mucopolisaccaridosi di tipo II (MPS II; OMIM #309900) (Fig. 6), nota anche come sindrome di Hunter, è il risultato di carenza dell’enzima iduronato-2-solfatasi (I2S) con conseguente accumulo di GAG di DS e HS . La prevalenza è di circa 1 su 140.000-156.000 nati vivi maschi . In particolare, questo è l’unico MPS X-linked, e le portatrici femminili sono solitamente asintomatiche; tuttavia, possono essere eccezionalmente colpite a causa di anomalie del cromosoma X, dell’omozigosità o dell’inattivazione distorta-X. L’espressione fenotipica abbraccia anche un ampio spettro di gravità clinica. Nei casi più gravi, un marcato coinvolgimento neurologico progressivo e una malattia somatica coesistono e la morte di solito si verifica nella seconda decade di vita. Altri pazienti con disfunzione neurologica minima o assente possono presentare un grave coinvolgimento somatico mentre, all’estremità opposta dello spettro, ci sono pazienti con solo minime manifestazioni somatiche e intelligenza normale che sopravvivono fino all’età adulta . I primi segni e sintomi sono condivisi dalle forme gravi di MPS I e II.

MPS II. Un paziente di 3 anni con solo lievi caratteristiche facciali caratteristiche
Casi sporadici di hydrops fetalis sono descritti in MPS I, anche se, in generale, questi bambini appaiono normali alla nascita. I primi segni di MPS I di solito compaiono durante i primi mesi di vita, mentre quelli in MPS II di solito si verificano un po ‘ più tardi . Kiely et al. , in uno studio retrospettivo in 55 pazienti di Hurler, ha osservato che i sintomi respiratori, difficoltà di alimentazione, ernia inguinale e otite media erano comparsi ad un’età mediana ≤ 6 mesi, e cifosi, anomalie cardiache, circonferenza cranica allargata, ipotonia, organomegalia, opacità corneale, restrizione articolare ed ernia ombelicale apparivano tra 6 e 12 mesi. L’ernia ombelicale è una delle caratteristiche presentanti più in anticipo in MPS I e II e le ernie inguinali sono riferite in circa 60% dei pazienti .
Un altro indizio iniziale comune per MPS I e II può essere infezioni ricorrenti delle vie aeree superiori con aumento delle secrezioni. L’otite frequente, la rinite cronica ricorrente e la secrezione nasale persistente senza infezione evidente non sono specifiche, ma possono aiutare a rafforzare il sospetto diagnostico se associate a segni più specifici. La conservazione del BAVAGLIO all’interno dell’oro-faringe porta all’allargamento delle tonsille e delle adenoidi e contribuisce alle complicanze delle vie aeree superiori. Un’altra scoperta frequente è la respirazione rumorosa, specialmente di notte, associata nelle fasi successive all’apnea ostruttiva del sonno. Come conseguenza di frequenti infezioni dell’orecchio medio e disostosi degli ossicini dell’orecchio medio, la perdita dell’udito è frequente, con deficit sia conduttivi che neurosensoriali . Tracheobronchomalacia è comunemente osservato e può portare a ostruzione acuta delle vie aeree o collasso, con questa è una delle principali cause di morte negli anni successivi .
Adenoidectomia, tonsillectomia e timpanostomia, insieme alla riparazione dell’ernia, sono gli interventi chirurgici più frequenti e precoci per i pazienti con MPS I e II, spesso eseguiti prima di conoscere la diagnosi (in oltre il 50% dei casi) .
La diarrea ricorrente è abbastanza comune nelle prime fasi di MPS I, e può essere presente anche nei pazienti affetti da MPS II con coinvolgimento neurologico, mentre è rara nei pazienti lievemente colpiti .
Deformità del gibbo (cifosi dorso-lombare) (Fig. 2) diventa clinicamente evidente ad un’età media di 8.6 mesi a 1 anno in MPS I, che rappresenta un marcatore abbastanza specifico e precoce di MPS in generale. I corpi vertebrali appaiono appiattiti e a becco, potenzialmente portando a complicazioni successive come intrappolamento del nervo spinale, lesione spinale acuta e instabilità atlanto-occipitale; pertanto, occorre prestare particolare attenzione per prevenire la dislocazione dell’articolazione atlanto-assiale durante l’anestesia generale e la chirurgia. Dopo 1 anno di età, contratture articolari e genu valgum rappresentano altri segni frequenti di compromissione articolare e displasia scheletrica progressiva, che sono facilmente rilevabili prima dei 2 anni di età in tutti i bambini affetti da MPS. Tutte queste manifestazioni scheletriche insieme, tipiche di MPS, sono chiamate multiplex di dysostosis. L’ispessimento delle costole può essere visto sulle radiografie (Fig. 3a), le ossa lunghe sono corte con alberi larghi e le ginocchia sono soggette a deformità in valgo e varo. La disostosi falangea e l’ispessimento sinoviale portano ad una caratteristica deformità della mano dell’artiglio (Fig. 4), che è un’altra caratteristica tipica che può sollevare il sospetto della malattia. La displasia dell’anca è spesso la ragione per cui questi bambini possono essere visti per la prima volta da specialisti ortopedici. Tipicamente, il bacino è scarsamente formato, le teste femorali sono piccole e la coxa valga è comune, con deformità progressiva e debilitante dell’anca (Fig. 3 ter).
C’è una notevole differenza tra MPS I e MPS II; in MPS I, la crescita scheletrica decelera di 3 anni di età , mentre i pazienti con MPS II mostrano una crescita eccessiva nei primi 5-6 anni di età, rallentando la loro crescita in seguito .
Ingrossamento del viso (naso largo con narici ampie, prominente sovraorbitario creste, ampia e arrotondata guance, labbra spesse, allargata sporgenti lingua) causato dal deposito di Gag nei tessuti molli della regione orofacciale e delle ossa facciali si manifesta entro i primi 2 anni, in un’età media di 1,1 anni per la malattia di Hurler e tra i 18 mesi e i 4 anni nella forma grave della malattia di Hunter , anche se sottili cambiamenti fenotipici può essere rilevato in precedenza (come già nel secondo semestre del primo anno) i pediatri con esperienza specifica (Fig. 1). La macrocefalia diventa progressivamente più evidente e la scafocefalia è comune (Fig. 5), ma la microcefalia è anche possibile nei pazienti con Hurler . L’irsutismo del viso e del corpo è sempre evidente all’età di 24 mesi . La pelle può essere ispessita e anelastica. In particolare, alcuni pazienti MPS II sviluppano una lesione cutanea distintiva, che è descritta come papule bianco-avorio (simili a ciottoli) sulla parte superiore della schiena e sui lati della parte superiore delle braccia, patognomonica della sindrome di Hunter .
La cardiomiopatia e il progressivo ispessimento e irrigidimento dei foglietti valvolari nei pazienti con MPS I possono portare a rigurgito mitralico e, meno frequentemente, aortico . Un soffio può essere rilevato nei primi mesi di vita e talvolta è stato segnalato come il primo segno della malattia . Il coinvolgimento cardiaco è presente anche in quasi tutti i pazienti con MPS II, ma questo inizia intorno ai 5 anni di età . La malattia della valvola può portare a ipertrofia ventricolare destra e sinistra e insufficienza cardiaca.
La protuberanza dell’addome causata da epatosplenomegalia progressiva è facilmente evidente dopo 6 mesi di età, sebbene la conservazione di GAG nel fegato e nella milza non porti a disfunzioni d’organo .
L’annebbiamento corneale si verifica in quasi tutti gli individui con MPS I prima dei 15 mesi di età , causando potenzialmente gravi disturbi visivi. Al contrario, l’annebbiamento corneale non è una caratteristica tipica della sindrome di Hunter , ma l’esame con lampada a fessura può rivelare lesioni corneali discrete che non influenzano la visione. La disfunzione retinica può essere rilevata, mentre il glaucoma non è una scoperta comune.
Lo sviluppo psicomotorio precoce è generalmente rilevato come normale, ma in MPS I il ritardo dello sviluppo è solitamente evidente all’età di 18 mesi, con un progressivo declino mentale che porta a una grave disabilità intellettiva. Le competenze linguistiche sono molto limitate in questi bambini. Un ritardo globale delle pietre miliari dello sviluppo in MPS II diventa evidente da 2 anni in poi . Tuttavia, la principale caratteristica neurologica della grave malattia di Hunter è rappresentata da disturbi comportamentali marcati, come iperattività, ostinazione e aggressività, che in genere non sono osservati in quelli con il fenotipo attenuato .
La mucopolisaccaridosi di tipo VII (MPS VII; OMIM #253220), nota anche come sindrome di Sly, è una MPS ultra-rara caratterizzata da attività carente di β-glucuronidasi, con accumulo lisosomiale di solfato di condroitina (CS), DS e HS, che porta a disfunzione cellulare e d’organo. Il primo paziente è stato descritto da Sly et al. nel 1973 e un totale di 145 pazienti sono stati riportati in letteratura fino ad ora.
La presentazione clinica e la progressione della malattia di MPS VII abbraccia un ampio spettro di gravità, da manifestazioni multisistemiche precoci, gravi e morte nei primissimi mesi, a un fenotipo più lieve con insorgenza successiva, intelligenza normale o quasi normale e sopravvivenza più lunga .
Le caratteristiche del fenotipo dei pazienti con MPS VII assomigliano a quelle di MPS I e MPS II (bassa statura, displasia scheletrica, macrocefalia, ipertrofia gengivale, infezioni ricorrenti dell’orecchio, epatosplenomegalia, ernie, coinvolgimento cardiaco, diminuzione della funzione polmonare e deterioramento cognitivo). Una percentuale inaspettatamente elevata di pazienti descritti (41%) ha una storia di hydrops fetalis neonatale non immune (NIHF) . Nonostante l’insorgenza prenatale della malattia in questi pazienti, 13 su 23 sono sopravvissuti all’infanzia con un decorso da lieve a intermedio nella tarda adolescenza. Pertanto, la presenza di NIHF non prevede, di per sé, la possibile gravità del decorso clinico se il paziente sopravvive all’infanzia. Un altro segno precoce è la macrocrania, mentre il coinvolgimento della valvola cardiaca e le infezioni ripetute del tratto respiratorio superiore e inferiore compaiono nei primi 2 anni di vita. Anche il ritardo mentale moderato e la progressiva perdita dell’udito con disturbi del linguaggio sono evidenti con il tempo.
In sintesi, le forme gravi di MPS I hanno un esordio multisistemico molto precoce (entro 6 mesi di vita); MPS II è simile ma con un esordio successivo, e MPS VII ha un esordio prenatale con frequente NIHF e precoce coinvolgimento grave del tratto respiratorio superiore e inferiore.
MPS con coinvolgimento principalmente neurologico e cognitivo
Mucopolisaccaridosi di tipo III (MPS III, nota anche come sindrome di Sanfilippo; Fig. 7) ha una presentazione neurologica prevalente. MPS III è un disturbo da accumulo lisosomiale causato dalla carenza di uno dei quattro enzimi coinvolti nel catabolismo dell’HS. Quattro diversi sottotipi sono noti—MPS III tipo A (OMIM #252900), di tipo B (OMIM #252920), tipo C (OMIM #252930), e di tipo D (OMIM #252940)—ad ogni effetto di un diverso deficit enzimatico: tipo A, sulfamidase/SGSH gene; tipo B, alfa-N-acetilglucosaminidasi/NAGLU gene; tipo C, alfa-glucosaminide N-acetiltransferasi/HGSNAT gene; tipo D, N-acetilglucosamina-6-solfato sulfatasi/GNS gene). Tutti e quattro i tipi (da A a D) hanno ereditarietà autosomica recessiva. MPS III è il più frequente dei MPS con una prevalenza stimata tra 0,3 e 4.1 casi per ogni 100.000 neonati, a seconda del sottotipo e della razza .

PARLAMENTARI IIIA. Lo stesso paziente a 2,5 e b 5 anni. Si noti la diversa espressione del viso, che mostra la progressione della compromissione neurologica
I pazienti con sindrome di Sanfilippo mostrano un progressivo deterioramento cognitivo nel secondo e terzo anno di vita. In contrasto con la maggior parte dei PARLAMENTARI, le caratteristiche somatiche sono relativamente meno evidenti e il coinvolgimento del SNC è cospicuo. Il ritardo dello sviluppo del linguaggio è di solito il primo segno notato dai genitori, ma le preoccupazioni principali possono essere problemi comportamentali (iperattività, comportamento ansioso e aggressivo, disturbi del sonno), seguiti da un progressivo declino mentale . Questi sintomi possono causare diagnosi errate come disturbi comportamentali, disturbo da deficit di attenzione e iperattività (ADHD) e disturbi dello spettro autistico . L’epilessia può anche essere presente.
I disturbi del sonno, la cui incidenza è riportata fino all ‘ 80-90%, sono una caratteristica comune. Consistono in difficoltà nell’addormentarsi e frequenti risvegli notturni, con completa inversione del ritmo giorno–notte in alcuni pazienti.
Tutte queste manifestazioni sono molto difficili da gestire e hanno un enorme impatto psicologico sulla qualità della vita per tutta la famiglia.
In generale, sebbene il fenotipo possa essere molto simile nei quattro sottotipi, il decorso clinico in Sanfilippo B e C sembra essere caratterizzato da un fenotipo meno grave .
I sintomi non neurologici sono di solito meno pronunciati in MPS III rispetto agli altri MPS. Le infezioni ricorrenti dell’orecchio, del naso e della gola sono osservate in età più giovane. Otite ricorrente, difetti ossicolari dell’orecchio medio e anomalie nell’orecchio interno possono causare sordità. Un eccesso di secrezioni spesse e cambiamenti anatomici possono produrre ostruzione delle vie aeree. Nei primi anni di vita, la diarrea è frequentemente riportata, mentre la stitichezza è più comune nei pazienti più anziani.
L’esame fisico può mostrare caratteristiche facciali grossolane, sebbene queste siano meno pronunciate. I pazienti hanno macrocefalia, ampie sopracciglia con svasamento mediale e sinofrisi, i capelli sono solitamente secchi e grossolani e l’ipertricosi è comune. Nei pazienti più giovani si osservano frequentemente epatomegalia lieve e ernie ombelicali e inguinali. Le contratture rare e spesso lievi si trovano principalmente nei gomiti. La crescita è influenzata in circa la metà dei pazienti di età superiore ai 12 anni .
In sintesi, la malattia MPS III o Sanfilippo ha un coinvolgimento neurologico prominente con segni somatici molto lievi. Bambini di età compresa tra 2-3 anni che sviluppano iperattività, ADHD e comportamento aggressivo possono essere sospettati di avere MPS III.
MPS con coinvolgimento somatico prevalente
MPS IV e MPS VI presenti con solo coinvolgimento somatico. Queste sono condizioni progressive che risparmiano la capacità intellettuale e colpiscono principalmente lo scheletro (MPS IVA), o tutti gli organi/sistemi del corpo (MPS VI). L’ereditarietà è autosomica recessiva. La velocità con cui i sintomi peggiorano varia tra gli individui affetti.
Mucopolisaccaridosi IV (MPS IV, nota anche come sindrome di Morquio; Fig. 8) si presenta come due geneticamente distinti disturbi, ognuna con un diverso deficit enzimatico: MPS IVA (sindrome di Morquio; OMIM #253000) a causa di carenza di N-acetilgalattosamina-6-solfatasi (GALNS gene), con conseguente accumulo di cheratan-solfato (KS) e CS ; e MPS IVB (Morquio B sindrome; OMIM #253010) a causa di deficit di beta-galattosidasi è leader di urina, KS e oligosaccaride escrezione come in GM1 pazienti. MPS IVB è infatti allelico alle varie forme di GM1-gangliosidosi, ma manca il deterioramento psicomotorio visto in GM1 gangliosidosi .

MPS IVA. 4 anno-vecchio paziente con b antero-posteriore e laterale c raggi x mostra “a forma di pala” costole e appiattito e arrotondato vertebre, con un tipico anteriore beaking” aspetto
i Pazienti con grave MPS IVA con non rilevabili attività dell’enzima rivelare i loro primi sintomi di solito durante il primo anno di vita, anche se sono raramente diagnosticato prima dei 4-5 anni di età. Le informazioni sulla storia naturale dei pazienti di Morquio A derivano principalmente da due ampie raccolte di dati . Il tasso di crescita ridotto inizia a circa 18 mesi di età e la crescita si fermerà a circa 7 o 8 anni di età. In contrasto con gli altri MPS, Morquio A articolazioni sono di solito lassista e molto flessibile (hypermobile), ma alcune articolazioni (di solito i fianchi e le spalle) possono avere una gamma limitata di movimento. I pazienti con tale coinvolgimento scheletrico possono ricevere una diagnosi di displasia spondiloepifisea, pseudoacondroplasia, displasia epifisaria multipla o malattia bilaterale di Legg-Calvé-Perthes .
Una caratteristica costante è l’ipoplasia odontoide; pertanto, la dislocazione dell’articolazione atlanto-occipitale può verificarsi causando compressione e danni al bulbare e al midollo spinale, con conseguente paralisi o addirittura morte se la stabilizzazione cervicale non viene eseguita precocemente.
I pazienti con MPS IVB hanno una manifestazione clinica molto simile ma con una bassa statura meno prominente. Hanno caratteristiche facciali leggermente grossolane, perdita dell’udito, opacità corneali, cardiopatia valvolare e frequenti infezioni del tratto respiratorio superiore. Possono anche presentare con ernia inguinale e lieve epatomegalia. Le caratteristiche displastiche scheletriche includono platispondilia, ipoplasia odontoide con possibile sublussazione cervicale, cifo-scoliosi, coxa valga e ali conache ristrette .
Mucopolisaccaridosi VI (MPS VI, nota anche come sindrome di Maroteaux-Lamy; OMIM #253200; Fig. 9) è dovuto a mutazioni patogene nel gene arilsulfatasi B (ARSB) situato sul cromosoma 5. Di conseguenza, l’attività ridotta o assente dell’enzima arilsulfatasi B (ASB) (N-acetilgalattosamina 4-solfatasi) compromette la degradazione del DS determinando il suo accumulo cellulare .

MPS VI. Un bambino di 2 anni con evidente faccia ruvida e disostosi scheletrica
I pazienti senza attività enzimatica rilevabile hanno la forma grave di MPS VI e presentano sintomi nella prima infanzia. Il loro fenotipo somatico è simile alla sindrome di Hurler. Nella maggior parte dei casi, entro 2 o 3 anni di età, diventa evidente un danno scheletrico grave e progressivo, chiamato dysostosis multiplex. Questa displasia scheletrica comprende displasia dell’anca con testa femorale displastica, corpi vertebrali anormali, clavicole irregolari, ulna distale e radio ipoplastiche e ossa metacarpali displastiche e corte; le ossa carpali e tarsali sono ipoplastiche e hanno un profilo irregolare. La velocità di crescita spesso rallenta dopo il primo anno di vita, con un’altezza finale generalmente inferiore a 120 cm. Come in altri PARLAMENTARI, un segno precoce è caratteristiche facciali grossolane. Inoltre, i pazienti hanno un tipico addome sporgente, epatomegalia, ernia ombelicale e/o inguinale e coinvolgimento cardiaco nella prima infanzia. Sebbene non sia presente alcuna disabilità intellettiva primaria, l’idrocefalo può essere osservato in alcuni pazienti con conseguente ipertensione intracranica e papilledema. Pectus carinatum, articolazioni rigide e contratte, scoliosi e cifosi e compressione del midollo spinale cervicale peggiorano con il tempo .
In sintesi, MPS IV e VI non mostrano coinvolgimento del SNC. MPS IV è un MPS unico nel mostrare lassità articolare, mentre l’insorgenza grave di MPS VI è simile a quella osservata in MPS I.