The genetics of cleft lip and palatal
 Intro/Abstractil labbro sinistro con o senza palatoschisi è un’anomalia congenita complessa che può essere isolata o vista insieme ad altre malformazioni. Può anche essere parte del fenotipo di una sindrome genetica. Questo articolo serve come una revisione della prevalenza di labbro leporino e palato, rischi di recidiva, e rischi per altre anomalie congenite. Saranno esplorate sindromi genetiche ed esposizioni teratogene note per essere associate a fessure orali. Inoltre, saranno discussi i test genetici comunemente richiesti nell’ambito della genetica clinica pediatrica per la valutazione del paziente con labbro leporino e palato.
Intro/Abstractil labbro sinistro con o senza palatoschisi è un’anomalia congenita complessa che può essere isolata o vista insieme ad altre malformazioni. Può anche essere parte del fenotipo di una sindrome genetica. Questo articolo serve come una revisione della prevalenza di labbro leporino e palato, rischi di recidiva, e rischi per altre anomalie congenite. Saranno esplorate sindromi genetiche ed esposizioni teratogene note per essere associate a fessure orali. Inoltre, saranno discussi i test genetici comunemente richiesti nell’ambito della genetica clinica pediatrica per la valutazione del paziente con labbro leporino e palato.
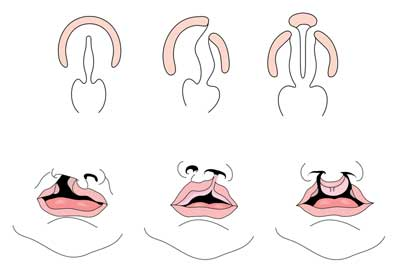
Labbro leporino con o senza palatoschisi (CL/CP) differisce da un palatoschisi isolato (CP) a livello embrionale, epidemiologico e genetico. Il labbro leporino deriva tipicamente dalla prominenza mascellare e dalla prominenza nasale mediale che non riescono a fondersi tra la quinta e la sesta settimana di sviluppo embrionale. Lo sviluppo normale del palato deriva dalla formazione del palato primario e del palato secondario. Il palato primario è formato alle settimane sei-sette dallo sviluppo e dalla fusione del nasale mediale, del nasale laterale e dei processi mascellari. Il palato secondario origina dalle mensole palatali (che si sviluppano dai processi mascellari accoppiati del primo arco branchiale) diventando orizzontale e fondendosi, formando i palati duri e molli intorno alla nona settimana di sviluppo embrionale. Gli scaffali si fondono anche con il palato primario e il setto nasale. (1)
Le fessure orali sono uno dei difetti alla nascita più comuni osservati nella scuola materna neonatale, con una prevalenza complessiva di 1.6 per mille neonati in tutto il mondo, con CL / CP visto in circa uno per mille nascite e CP visto in 0.6 per mille nascite. (2) C’è una maggiore frequenza di CL/CP in individui di origine asiatica, africana e nativa americana. CL / CP è anche più comune nei maschi. Al contrario, non vi è alcuna differenza significativa nell’incidenza di CP tra diversi background etnici, e CP è più comune nelle donne. (3) I rischi di recidiva all’interno di una famiglia dipendono dal fatto che la fessura sia isolata (senza altri risultati clinici presenti) o vista come parte di una sindrome genetica. La maggior parte dei casi di fessure orali sono isolati (circa l ‘ 80%). Si ritiene che le fessure isolate abbiano un’eredità multifattoriale: sono dovute a una combinazione di molteplici fattori, sia genetici che ambientali. Il rischio di recidiva (Tabella 1) aumenta quando vi è più di un parente affetto. Il rischio di recidiva aumenta anche più grave è il difetto.

Labbro leporino e palato può essere visto con altre anomalie congenite. La probabilità di un’eziologia genetica o teratogena aumenta più anomalie congenite con cui un paziente presenta. La presenza di altri problemi come la disabilità intellettiva, problemi comportamentali come l’autismo, caratteristiche dismorfiche o altre preoccupazioni mediche renderà anche più probabile una malattia genetica o un’esposizione teratogena. Circa il 13% degli individui con labbro leporino avrà altre preoccupazioni mediche o anomalie. Il numero aumenta al 37% con labbro leporino e palato e al 47% con palatoschisi da solo.
L’esposizione prenatale ad agenti teratogeni (come talidomide, anticonvulsivanti, alcol, acido retinoico e sigarette) e malattie materne (come diabete, rosolia e carenza di folati) hanno dimostrato di aumentare il rischio di fessure orali. La presenza di bande amniotiche aumenta anche il rischio di fessure. È noto che la supplementazione di acido folico periconceptuale riduce il rischio di fessure orali.
La sequenza di Pierre Robin è un’anomalia craniofacciale caratterizzata da ipoplasia mandibolare o micrognazia, palatoschisi secondaria a forma di U e glossoptosi che porta ad apnea ostruttiva e difficoltà di alimentazione. La sequenza di Pierre Robin può essere vista come parte delle sindromi genetiche (sindrome da delezione 22q11.2, sindrome di Stickler; descritta di seguito). (5)
Ci sono centinaia di sindromi genetiche associate a fessure orali, incluse anomalie citogenetiche (aneuploidie, microdelezioni) e disturbi a singolo gene (Mendeliano). Confermare una diagnosi genetica è essenziale per determinare la prognosi e stabilire un rischio di recidiva.
Le aneuploidie come la trisomia 13 e 18 hanno una forte associazione con CL / CP. La trisomia 13 (AKA sindrome di Patau) è associata a tre copie del cromosoma 13, o traslocazioni Robertsoniane sbilanciate che coinvolgono il cromosoma 13. I bambini nati con questa circostanza muoiono tipicamente nel periodo neonatale. Le caratteristiche cliniche includono labbro leporino e palato, ritardo della crescita, gravi malformazioni del sistema nervoso centrale (inclusa l’oloprosencefalia), microcefalia, microptalmia, coloboma dell’iride, assenza degli occhi, orecchie malformate, polidattilia, pugni serrati, piedi inferiori a bilanciere, difetti cardiaci congeniti e difetti urogenitali. Le fessure della linea mediana (altrimenti molto rare) possono essere osservate nella trisomia 13 a causa del rischio di difetti della linea mediana, inclusa l’oloprosencefalia. La trisomia 18 (sindrome di Edwards di AKA) è dovuta tipicamente a tre copie distinte del cromosoma 18 ed è associata con l’esito postnatale povero. Le caratteristiche cliniche includono labbro leporino e palato, disabilità intellettiva, incapacità di prosperare, cardiopatia congenita, ipertonia, micrognatia, sterno corto, orecchie malformate basse, mani serrate, piedi inferiori a bilanciere e unghie ipoplastiche, tra gli altri. La trisomia 13 e 18 può essere facilmente confermata o esclusa facendo analisi cromosomiche (cariotipo).
Le sindromi da microdelezione comportano tipicamente la delezione di parte di un cromosoma. Queste eliminazioni possono essere troppo piccole per essere rilevate mediante cariotipo standard e possono richiedere l’individuazione della tecnologia FISH (fluorescenza in situ ibridazione) o microarray. Una ben nota sindrome da microdelezione associata alla palatoschisi è la sindrome da delezione 22q11.2 (aka Digeorge/sindrome velocardiofacciale). Anomalie palatali tra cui incompetenza velofaringea, fessure sottomucose, uvula bifida e palatoschisi sono osservate nel 69% degli individui con delezione 22q11.2 e possono far parte della sequenza Pierre Robin. Altri risultati clinici includono cardiopatia congenita, perdita dell’udito, caratteristiche dismorfiche, immunodeficienza, ipocalcemia, anomalie renali, problemi di alimentazione, anomalie scheletriche e disturbi psichiatrici. Si ritiene che circa il 10% dei casi di sindrome da delezione 22q11.2 sia familiare. La delezione si segrega in modo autosomico dominante.(6) La sindrome di Wolf-Hirschhorn, che è dovuta a una delezione nel braccio corto del cromosoma 4, è anche associata a fessure orali (nel 25% al 50% degli individui affetti). Caratteristiche facciali caratteristiche (tra cui glabella prominente che porta a “aspetto casco greco-guerriero”), cardiopatia congenita, disabilità intellettiva, convulsioni, incapacità di prosperare, micrognazia, tag preauricolari o pozzi e ipodontia possono anche essere visti come parte della condizione.(7)
I disturbi monogenici con fessure orali includono la sindrome di Stickler, la sindrome di Treacher Collins e la sindrome di Van der Woude, tra molti altri. La sindrome di Stickler è una malattia del collagene con ereditarietà autosomica dominante e, meno comunemente, autosomica recessiva. Caratteristiche comuni includono palatoschisi (visto come parte della sequenza di Pierre Robin o senza micrognazia), perdita dell’udito (neurosensoriale e conduttiva), reperti scheletrici (artrite precoce, displasia spondiloepifisea), anomalie oculari (alta miopia, anomalie vitree) e caratteristiche facciali caratteristiche (con sottosviluppo della mascella e del ponte nasale, retrusione midface). I test genetici per la sindrome di Stickler possono essere complessi, poiché sono state descritte mutazioni in almeno sei geni in individui affetti. Circa il 90% dei pazienti con sindrome di Stickler ha mutazioni nel gene COL2A1 e ha una forma autosomica dominante della condizione.(8) La sindrome di Treacher Collins è una condizione autosomica dominante caratterizzata da palatoschisi con o senza labbro leporino nel 28% degli individui affetti. Altre anomalie includono ipoplasia delle ossa zigomatiche e della mandibola, anomalie dell’orecchio esterno, coloboma della palpebra inferiore, perdita dell’udito conduttivo, assenza di ciglia inferiori, spostamento preauricolare dei capelli sulle guance e stenosi coanale o atresia. La diagnosi della sindrome di Treacher Collins si basa su risultati clinici e radiografici. Sono state descritte mutazioni in almeno tre geni, con mutazioni nel TCOF1 osservate nel 78% -93% dei pazienti.(9) La sindrome di Van der Woude è caratterizzata dalla presenza di fistole congenite, di solito bilaterali, paramediane del labbro inferiore (fosse), o talvolta piccoli tumuli con un tratto sinusale che porta da una ghiandola mucosa del labbro e fessure orali (tra cui CL/CP e CP). Van der Woude è una condizione autosomica dominante associata a mutazioni nel gene IRF6 (10). Il test per le condizioni a singolo gene o multi-gene richiede un’analisi diretta del gene mediante sequenziamento e/o analisi di delezione / duplicazione (come MLPA).
Dato che le sindromi genetiche con labbro leporino e palato possono essere associate a aneuploidie, microdelezioni/microduplicazioni cromosomiche o disturbi monogenici, i test genetici possono essere un processo complicato. Una storia medica approfondita, un pedigree di tre generazioni, una storia di gravidanza e un esame di dismorfologia da parte di un genetista clinico possono chiarire il quadro clinico e consentire test genetici mirati. Le tecnologie più recenti, tra cui microarray, consentiranno l’identificazione di piccole microdelezioni e microduplicazioni precedentemente mancate dal cariotipo standard. Sfortunatamente, questa tecnica porta anche all’identificazione di delezioni e duplicazioni di significato clinico sconosciuto, complicando il processo di consulenza genetica. Il test per i disturbi monogenici o i disturbi mendeliani richiede la disponibilità clinica di test genetici per il gene desiderato. Può anche essere costoso se non coperto da assicurazione medica. Nuove tecnologie come il sequenziamento di nuova generazione, il sequenziamento degli esomi o il sequenziamento del genoma (noti collettivamente come test genomici) sono ora diventati clinicamente disponibili. Analizzando centinaia a migliaia di geni contemporaneamente, questi test aumentano significativamente la potenza diagnostica e la resa. Rispetto ad altre tecniche, questi test possono fornire una risposta più veloce e in modo più economico. Nel campo della ricerca, il sequenziamento dell’esoma e del genoma ha portato all’identificazione di nuovi geni e all’espansione delle caratteristiche cliniche e dello spettro per le mutazioni genetiche. Come con la tecnologia microarray, i test genomici possono rilevare sindromi che non sono correlate alla presentazione del paziente e/o alla ragione del test. Data la complessità intrinseca dei test genetici, è necessario il consenso informato.
Conclusione
Sebbene il labbro leporino e il palato siano un’anomalia isolata nella maggior parte dei casi, esiste una forte associazione tra le fessure orali e altre anomalie e sindromi genetiche. Una valutazione genetica da parte di un genetista clinico e di un consulente genetico è essenziale per la guida anticipatoria e per determinare i rischi di recidiva. I test genetici, che richiedono il consenso informato, possono essere coordinati e interpretati durante una valutazione genetica.
Anya Revah, MS, è il consulente genetico senior presso la Divisione di genetica medica presso Maimonides Infants and Children’s Hospital di Brooklyn, New York. Lei è anche un membro attivo del Maimonides Medical Center e Kings County Hospital Labbro leporino e palato Team multidisciplinare. Ha un Master in Scienze in Consulenza genetica presso la Boston University di Boston, Massachusetts.
1. Sadler TW. L’Embriologia medica di Langman. Nona edizione. Pagine 390-395.
2. Per la prevenzione dei difetti di nascita della rete nazionale, Parker SE, Mai CT, Canfield MA, Rickard R, Wang Y, Meyer RE, Anderson P, Mason CA, Collins JS, Kirby RS, Correa A. Aggiornate le stime nazionali di prevalenza di nascita per difetti alla nascita selezionati negli Stati Uniti. 2004-2006. Ricerca sui difetti alla nascita (Parte A): Teratologia clinica e molecolare 2010;88: 1008-1016.
3. Fraser FC. La genetica di labbro leporino e palatoschisi. Essere. J. Hum. Genet. 1970;22: 336–352.
4. Van Rooij IA, Ocke MC, et al. L’assunzione di folato periconceptuale mediante integratore e assunzione di cibo riduce il rischio di labbro leporino non sindromico con o senza palatoschisi. Prev Med 2004; 39: 689-694.
5. Tan TY. Kilpatrick N, Farlie PG. Prospettive di sviluppo e genetiche sulla sequenza di Pierre Robin. Essere. J. Med. Genet. 2013;163C: 295-305.
6. McDonald-McGinn DM, Emanuel BS, Zackai EH. 22q11. 2 Sindrome da delezione. Settembre 23, 1999. . In: Pagon RA, Adam MP, Ardinger HH, et al., editore. GeneReviews . Seattle (WA): Università di Washington, Seattle; 1993-2014. Disponibile a partire da: http://www.ncbi.nlm.nih.gov/books/NBK1523/.
7. Battaglia A, Carey JC, South ST, et al. Sindrome di Wolf-Hirschhorn. Apr. 29, 2002. . In: Pagon RA, Adam MP, Ardinger HH, et al., editore. GeneReviews . Seattle (WA): Università di Washington, Seattle; 1993-2014. Disponibile presso: http://www.ncbi.nlm.nih.gov/books/NBK1183/.
8. Robin NH, Moran RT, Ala-Kokko L. Sindrome di Stickler. Jun. 9, 2000. . In: Pagon RA, Adam MP, Ardinger HH, et al., editore. GeneReviews . Seattle (WA): Università di Washington, Seattle; 1993-2014. Disponibile presso: http://www.ncbi.nlm.nih.gov/books/NBK1302/.
9. Katsanis SH, Jabs EW. Sindrome di Treacher Collins. Lug. 20, 2004. . In: Pagon RA, Adam MP, Ardinger HH, et al., editore. GeneReviews . Seattle (WA): Università di Washington, Seattle; 1993-2014. Disponibile presso: http://www.ncbi.nlm.nih.gov/books/NBK1532/.
10. Schutte BC, Saal HM, Goudy S, et al. Disturbi correlati all’IRF6. Oct. 30, 2003. . In: Pagon RA, Adam MP, Ardinger HH, et al., editore. GeneReviews . Seattle (WA): Università di Washington, Seattle; 1993-2014. Disponibile presso: http://www.ncbi.nlm.nih.gov/books/NBK1407/.