Mucopolysaccharidosen: frühe diagnostische Anzeichen bei Säuglingen und Kindern
Mucopolysaccharidosen (MPS) sind eine Gruppe klinisch heterogener Erkrankungen, die durch Mängel der lysosomalen Enzyme verursacht werden, die für den Abbau von Glykosaminoglykanen (GAGs) erforderlich sind.
MPS sind durch progressive und systemische klinische Manifestationen gekennzeichnet. Trotz ihrer biochemischen und genetischen Heterogenität teilen verschiedene Typen wichtige klinische Merkmale in unterschiedlichen Kombinationen, einschließlich Gelenk- und Skelettdysplasie mit Steifheit (außer MPS IV, wo es Laxheit gibt) und Schmerzen, grobe Gesichtszüge, Hornhauttrübung, Leisten- oder Bauchhernien, wiederkehrende Infektionen der oberen Atemwege, Herzklappenerkrankungen, Karpaltunnelsyndrom und variable neurologische Beteiligung. Diese Merkmale treten normalerweise in den ersten Lebensmonaten für die schweren Formen und in der frühen Kindheit für die abgeschwächten Formen auf, werden jedoch oft unterschätzt und im Allgemeinen nur dann berücksichtigt, wenn sie deutlich erkennbar sind.
Die Seltenheit dieser Störungen und die Variabilität des klinischen Erscheinungsbildes führen häufig zu einer Verzögerung der Diagnose; Dies kann bei den schweren Formen von Monaten bis zu Jahren bei den abgeschwächten Formen reichen (siehe Rigoldi et al. in dieser Ergänzung ). Aufgrund des schnellen Fortschreitens und der dringenden Notwendigkeit eines Eingriffs in die schwere Phase kann jedoch selbst eine monatelange Verzögerung der Diagnose zu katastrophalen Ergebnissen für die zukünftige Gesundheit des Patienten führen.
Unser Ziel ist es, in dieser Übersicht die sehr frühen Anzeichen der schweren Formen hervorzuheben, die den Kinderarzt bei ihrem ersten Auftreten alarmieren sollten.
- Welche Anzeichen / Symptome können wir bei Kindern mit schweren Formen von MPS erwarten?
- Was sind die verschiedenen Darstellungen der verschiedenen Arten von MPS?
- MPS mit somatischer und kognitiver Beteiligung
- MPS mit überwiegend neurologischer und kognitiver Beteiligung
- MPS mit vorherrschender somatischer Beteiligung
Welche Anzeichen / Symptome können wir bei Kindern mit schweren Formen von MPS erwarten?
Symptome und Anzeichen, die auf verschiedene Formen von MPS hindeuten, sind in Tabelle 1 nach Alter angegeben. In den ersten 6 Lebensmonaten können bei MPS-Patienten Leistenhernien, ungewöhnlich häufige Atemwegsinfektionen, Otitis und Organomegalie beobachtet werden, insbesondere bei MPS I (Hurler-Syndrom) mit dem frühesten schweren Beginn. Darüber hinaus entwickeln diese Säuglinge zwischen 6 und 12 Monaten sehr häufig einen Gibbus (toraco-lumbale Kyphose), Herzgeräusch, Nabelbruch, leichte Hypotonie und Wachstumsverzögerung . Sie können auch das typische grobe Gesichtsausdruck (Abb. 1). MPS II, VI und VII können einen ähnlichen frühen Phänotyp aufweisen. MPS IV-Patienten haben einen frühen Skelettphänotyp mit dem Auftreten von Hüftdysplasie in den ersten Lebensmonaten und Taubenbrust (Pectus carinatum) in den ersten 12-18 Monaten. MPS II-Säuglinge weisen eine ähnliche Beteiligung auf, treten jedoch später auf. Im zweiten Lebensjahr entwickeln Säuglinge mit MPS I-Hurler-Syndrom eine kognitive Verzögerung, während ein MPS II-Patient möglicherweise nur aufgrund von Überwucherung, häufigen Atemwegsinfektionen und mehreren Operationen identifiziert wird ; Die typischen Gesichtszüge treten erst später auf. Einige MPS-II-Patienten entwickeln auch Hyperaktivität zwischen 18 und 24 Monaten. Im dritten Lebensjahr sind Hyperaktivität und kognitive Verzögerung bei MPS II und MPS III leicht erkennbar.
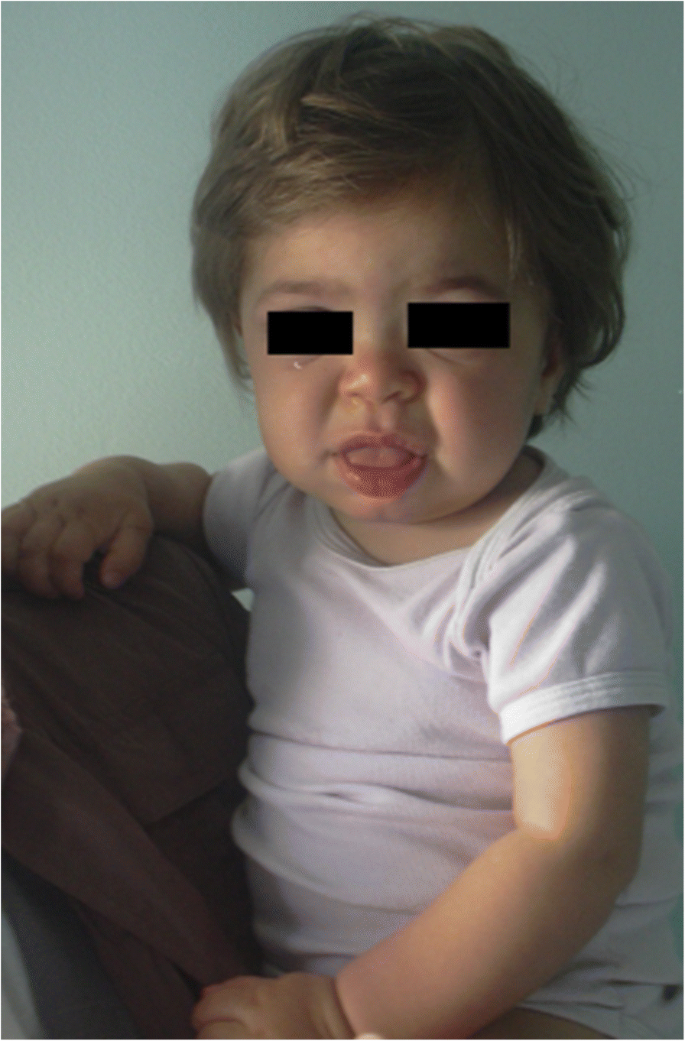
MPS IH bei 1-jährigem Patienten. Grobe Fazies: flache Nasenbrücke, Makroglossie, frontales Bossing
Zusammenfassend sind Skelettmanifestationen typisch und manchmal frühreif. Grobe Fazies und kognitive Verzögerung sind keine frühen auffälligen Anzeichen.
Was sind die verschiedenen Darstellungen der verschiedenen Arten von MPS?
MPS mit somatischer und kognitiver Beteiligung
Mukopolysaccharidose Typ I (MPS I; OMIM #252800) (Abb. 1, 2, 3, 4 und 5) wird durch einen Mangel der lysosomalen Hydrolase α-l-Iduronidase verursacht, was zu einer Multisystemakkumulation von zwei GAGs führt: Dermatansulfat (DS) und Heparansulfat (HS) . Basierend auf seinem Schweregrad wird MPS I traditionell in drei klinische Phänotypen unterteilt; Diese Formen stellen jedoch ein Kontinuum der Schwere der Erkrankung dar. Das Hurler-Syndrom, die schwerste Form, tritt typischerweise im ersten Lebensjahr auf. Betroffene Kinder entwickeln schnell signifikante kognitive Beeinträchtigungen und somatische Erkrankungen in mehreren Organsystemen, die innerhalb des ersten Jahrzehnts ohne Behandlung zum Tod führen. Die abgeschwächten Formen von MPS I, bekannt als Hurler-Scheie-Syndrom bzw. Scheie-Syndrom, sind durch späteres Auftreten von Symptomen, längere Lebenserwartung und leichte oder keine Beteiligung des Zentralnervensystems (ZNS) gekennzeichnet . Die Vererbung ist autosomal-rezessiv, und in mindestens 50% der Fälle ist eine Phänotypvorhersage auf der Grundlage des Genotyps möglich, während in den verbleibenden 50% neue Mutationen nachgewiesen werden . Die Prävalenz liegt bei etwa 1 von 100.000 Lebendgeburten .

MPS IH. ein Gibbus bei einem 9 Monate alten Patienten. b Wirbelsäule Röntgenaufnahme der Wirbelsäule bei einem 3-jährigen Patienten. c T2-gewichtete Magnetresonanzaufnahme der Wirbelsäule mit signifikanter Kyphose und Wirbelkanalstenose

MPS IH. Röntgennachweis von Dysostose Multiplex. eine Verdickung der Rippen bei einem 3-jährigen Patienten und eine Hüftdysplasie bei einem anderen Patienten im Alter von 18 Monaten

MPS IH: eine Klauenhand bei einem 18 Monate alten Patienten; b Hand Röntgen.

MPS IH. eine Verdickung des kortikalen Schädelknochens und eine abnormale “J-förmige” Konformation der Sella turcica. Dilatation der periventrikulären Räume in b FLAIR und c T-2 gewichteten Magnetresonanzbildern eines Patienten im Alter von 17 Monaten
Mukopolysaccharidose Typ II (MPS II; OMIM #309900) (Abb. 6), auch Hunter-Syndrom genannt, ist das Ergebnis eines Mangels des Enzyms Iduronat-2-sulfatase (I2S) mit anschließender GAG-Akkumulation von DS und HS . Die Prävalenz liegt bei 1 von 140.000 bis 156.000 männlichen Lebendgeburten . Bemerkenswerterweise ist dies die einzige X-chromosomale MPS, und weibliche Träger sind in der Regel asymptomatisch; Sie können jedoch aufgrund von Anomalien des X-Chromosoms, Homozygotie oder Schiefe-X-Inaktivierung ausnahmsweise betroffen sein . Die phänotypische Expression umfasst auch ein breites Spektrum klinischer Schweregrade. In schweren Fällen koexistieren eine ausgeprägte progressive neurologische Beteiligung und eine somatische Erkrankung, und der Tod tritt normalerweise im zweiten Lebensjahrzehnt auf. Andere Patienten mit minimaler oder keiner neurologischen Dysfunktion können eine schwere somatische Beteiligung aufweisen, während es am anderen Ende des Spektrums Patienten mit nur minimalen somatischen Manifestationen und normaler Intelligenz gibt, die bis ins Erwachsenenalter überleben . Frühe Anzeichen und Symptome werden von den schweren Formen von MPS I und II geteilt.

MPS II. Ein 3-jähriger Patient mit nur leichten charakteristischen Gesichtszügen
Sporadische Fälle von Hydrops fetalis sind in MPS I beschrieben , obwohl diese Kinder im Allgemeinen bei der Geburt normal erscheinen. Die ersten Anzeichen von MPS I treten normalerweise in den ersten Lebensmonaten auf, während die von MPS II normalerweise etwas später auftreten . Kiely et al. , in einer retrospektiven Studie an 55 Hurler-Patienten, beobachtet, dass respiratorische Symptome, Fütterungsschwierigkeiten, Leistenbruch und Mittelohrentzündung im mittleren Alter ≤ 6 Monate aufgetreten waren und Kyphose, Herzanomalien, vergrößerter Kopfumfang, Hypotonie, Organomegalie, Hornhauttrübung, Gelenkbeschränkung und Nabelbruch zwischen 6 und 12 Monaten auftraten. Nabelhernie ist eines der frühesten Merkmale bei MPS I und II, und Leistenhernien werden bei etwa 60% der Patienten berichtet .
Ein weiterer häufiger erster Hinweis auf MPS I und II können rezidivierende Infektionen der oberen Atemwege mit erhöhten Sekreten sein. Häufige Otitis, chronisch rezidivierende Rhinitis und anhaltender Nasenausfluss ohne offensichtliche Infektion sind nicht spezifisch, können jedoch dazu beitragen, den diagnostischen Verdacht zu verstärken, wenn sie mit spezifischeren Anzeichen verbunden sind. Die Lagerung von GAG im Oropharynx führt zu einer Vergrößerung der Mandeln und Polypen und trägt zu Komplikationen der oberen Atemwege bei. Ein weiterer häufiger Befund ist lautes Atmen, besonders nachts, verbunden in den späteren Stadien mit obstruktiver Schlafapnoe. Als Folge häufiger Mittelohrentzündungen und Dysostosen der Gehörknöchelchen des Mittelohrs kommt es häufig zu Hörverlust mit leitenden und sensorineuralen Defiziten . Tracheobronchomalazie wird häufig beobachtet und kann zu einer akuten Atemwegsobstruktion oder einem Kollaps führen, wobei dies eine der Haupttodesursachen in den folgenden Jahren ist .
Adenoidektomie, Tonsillektomie und Tympanostomie sind zusammen mit der Hernienreparatur die häufigsten und frühesten chirurgischen Eingriffe bei Patienten mit MPS I und II, die häufig vor Kenntnis der Diagnose durchgeführt werden (in mehr als 50% der Fälle) .
Rezidivierende Diarrhöe ist in frühen Stadien von MPS I recht häufig und kann auch bei MPS II-Patienten mit neurologischer Beteiligung auftreten, während sie bei leicht betroffenen Patienten ungewöhnlich ist .
Gibbusdeformität (dorso-lumbale Kyphose) (Abb. 2) wird im mittleren Alter von 8 Jahren klinisch sichtbar.6 Monate bis 1 Jahr in MPS I , was einen ganz spezifischen und frühreifen Marker für MPS im Allgemeinen darstellt. Wirbelkörper erscheinen abgeflacht und Schnabel, was möglicherweise zu späteren Komplikationen wie Spinalnerveneinklemmung, akute Wirbelsäulenverletzung, und Atlanto-Occipital-Instabilität; so, Besondere Vorsicht ist geboten, um eine Dislokation des Atlanto-Axialgelenks während der Vollnarkose und Operation zu verhindern. Nach dem Alter von 1 Jahr stellen Gelenkkontrakturen und Genu Valgum weitere häufige Anzeichen einer Gelenkbeeinträchtigung und einer fortschreitenden Skelettdysplasie dar, die bei allen von MPS betroffenen Säuglingen bereits im Alter von 2 Jahren leicht nachweisbar sind. Alle diese für MPS typischen Skelettmanifestationen zusammen werden als Dysostose-Multiplex bezeichnet. Eine Verdickung der Rippen ist auf Röntgenbildern zu sehen (Abb. 3a) sind die langen Knochen kurz mit breiten Schäften und die Knie neigen zu Valgus- und Varusdeformitäten. Phalangeale Dysostose und Synovialverdickung führen zu einer charakteristischen Klauenhand-Deformität (Abb. 4), was ein weiteres typisches Merkmal ist, das den Verdacht auf die Krankheit erwecken kann. Hüftdysplasie ist häufig der Grund, warum diese Kinder zuerst von Orthopäden gesehen werden können. Typischerweise ist das Becken schlecht geformt, die Femurköpfe sind klein und Coxa valga ist häufig mit fortschreitender und schwächender Hüftdeformität (Abb. 3b) .
Es gibt einen bemerkenswerten Unterschied zwischen MPS I und MPS II; Bei MPS I verlangsamt sich das Skelettwachstum um 3 Jahre , während MPS II-Patienten in den ersten 5-6 Jahren ein Überwachsen zeigen und danach ihr Wachstum verlangsamen .
Eine Vergröberung der Gesichtszüge (breite Nase mit ausgestellten Nasenlöchern, markante supraorbitale Grate, große abgerundete Wangen, dicke Lippen, vergrößerte hervorstehende Zunge), die durch die Speicherung von GAGs in den Weichteilen der orofazialen Region und der Gesichtsknochen verursacht wird, zeigt sich innerhalb der ersten 2 Jahre, bei einem Durchschnittsalter von 1, 1 Jahren bei der Hurler-Krankheit und zwischen 18 Monaten und 4 Jahren bei der schweren Form der Hunter-Krankheit , obwohl subtile phänotypische Veränderungen früher erkannt werden können (bereits in der zweiten Hälfte des erstes Jahr) von Kinderärzten mit spezifischer Erfahrung (Abb. 1). Makrozephalie wird zunehmend offensichtlicher und Scaphocefaly ist häufig (Abb. 5), aber Mikrozephalie ist auch bei Hurler-Patienten möglich . Gesichts- und Körperhirsutismus sind immer im Alter von 24 Monaten erkennbar . Die Haut kann verdickt und unelastisch sein. Insbesondere entwickeln einige MPS II-Patienten eine ausgeprägte Hautläsion, die als elfenbeinweiße Papeln (kieselartig) am oberen Rücken und an den Seiten der Oberarme beschrieben wird, pathognomonisch für das Hunter-Syndrom .
Kardiomyopathie und fortschreitende Verdickung und Versteifung der Klappenblätter bei MPS I-Patienten können zu Mitral- und seltener zu Aorteninsuffizienz führen . Ein Murmeln kann in den ersten Lebensmonaten festgestellt werden und kann manchmal als erstes Anzeichen der Krankheit gemeldet werden . Eine Herzbeteiligung ist auch bei fast allen Patienten mit MPS II vorhanden, dies beginnt jedoch im Alter von etwa 5 Jahren . Eine Klappenerkrankung kann zu rechts- und linksventrikulärer Hypertrophie und Herzinsuffizienz führen.
Die durch fortschreitende Hepatosplenomegalie verursachte Protuberanz des Abdomens ist nach 6 Monaten leicht erkennbar, obwohl die Speicherung von GAGs in Leber und Milz nicht zu einer Organfunktionsstörung führt .
Hornhauttrübung tritt bei fast allen Personen mit MPS I im Alter von weniger als 15 Monaten auf und kann zu schweren Sehstörungen führen. Im Gegenteil, Hornhauttrübung ist kein typisches Merkmal des Hunter-Syndroms , aber die Spaltlampenuntersuchung kann diskrete Hornhautläsionen aufdecken, die das Sehvermögen nicht beeinträchtigen. Eine Netzhautfunktionsstörung kann festgestellt werden, während ein Glaukom kein häufiger Befund ist.
Die frühe psychomotorische Entwicklung wird im Allgemeinen als normal erkannt, aber bei MPS I ist die Entwicklungsverzögerung normalerweise im Alter von 18 Monaten offensichtlich, wobei ein fortschreitender geistiger Verfall zu einer schweren geistigen Behinderung führt. Die Sprachkenntnisse sind bei diesen Kindern sehr begrenzt. Eine globale Verzögerung der Entwicklungsmeilensteine in MPS II wird ab 2 Jahren deutlich . Das neurologische Hauptmerkmal der schweren Hunter-Krankheit sind jedoch ausgeprägte Verhaltensstörungen wie Hyperaktivität, Hartnäckigkeit und Aggressivität, die bei Patienten mit dem abgeschwächten Phänotyp typischerweise nicht beobachtet werden .
Mucopolysaccharidose Typ VII (MPS VII; OMIM #253220), auch als Sly-Syndrom bekannt, ist eine extrem seltene MPS, die durch eine mangelhafte Aktivität der β-Glucuronidase mit lysosomaler Speicherung von Chondroitinsulfat (CS), DS und HS gekennzeichnet ist und zu Zell- und Organfunktionsstörungen führt. Der erste Patient wurde von Sly et al. im Jahr 1973 und insgesamt 145 Patienten wurden bisher in der Literatur berichtet.
Das klinische Erscheinungsbild und der Krankheitsverlauf von MPS VII umfassen ein breites Schweregradspektrum, von frühen, schweren Multisystemmanifestationen und Tod in den ersten Monaten bis hin zu einem milderen Phänotyp mit späterem Beginn, normaler oder nahezu normaler Intelligenz und längerem Überleben .
Phänotypische Merkmale von Patienten mit MPS VII ähneln denen von MPS I und MPS II (Kleinwuchs, Skelettdysplasie, Makrozephalie, Zahnfleischhypertrophie, wiederkehrende Ohrinfektionen, Hepatosplenomegalie, Hernien, Herzbeteiligung, verminderte Lungenfunktion und kognitive Beeinträchtigung). Ein unerwartet hoher Anteil der beschriebenen Patienten (41%) hat eine Vorgeschichte von nicht-immunem neonatalem Hydrops fetalis (NIHF) . Trotz des pränatalen Ausbruchs der Krankheit bei diesen Patienten überlebten 13 von 23 die Kindheit mit einem leichten bis mittleren Verlauf bis ins späte Teenageralter. Daher sagt das Vorhandensein von NIHF an sich nicht die mögliche Schwere des klinischen Verlaufs voraus, wenn der Patient die Kindheit überlebt. Ein weiteres frühes Zeichen ist Makrokranie, während Herzklappenbeteiligung und wiederholte Infektionen der oberen und unteren Atemwege in den ersten 2 Lebensjahren auftreten. Moderate geistige Behinderung und fortschreitender Hörverlust mit Sprachstörungen sind ebenfalls mit der Zeit offensichtlich.
Zusammenfassend haben die schweren Formen von MPS I einen sehr frühen Multisystem-Beginn (innerhalb von 6 Lebensmonaten); MPS II ist ähnlich, aber mit einem späteren Beginn, und MPS VII hat einen pränatalen Beginn mit häufigen NIHF und frühen schweren Beteiligung der oberen und unteren Atemwege.
MPS mit überwiegend neurologischer und kognitiver Beteiligung
Mucopolysaccharidosen Typ III (MPS III, auch bekannt als Sanfilippo-Syndrom; Abb. 7) hat eine vorherrschende neurologische Präsentation. MPS III ist eine lysosomale Speicherstörung, die durch einen Mangel an einem der vier Enzyme verursacht wird, die am Katabolismus von HS beteiligt sind. Es sind vier verschiedene Subtypen bekannt — MPS III Typ A (OMIM # 252900), Typ B (OMIM # 252920), Typ C (OMIM # 252930) und Typ D (OMIM # 252940) — jeweils aufgrund eines anderen Enzymmangels: Typ A, Sulfamidase / SGSH-Gen; Typ B, Alfa-N-Acetylglucosaminidase / NAGLU-Gen; Typ C, Alfa-Glucosaminid-N-Acetyltransferase / HGSNAT-Gen ; typ D, N-acetilglucosamina-6-solfato sulfatasi/GNS-Gen). Alle vier Typen (A bis D) sind autosomal-rezessiv vererbt. MPS III ist die häufigste der MPS mit einer geschätzten Prävalenz zwischen 0,3 und 4.1 Fälle pro 100.000 Neugeborene, abhängig von Subtyp und Rasse .

MPS IIIA. Der gleiche Patient bei a 2,5 und b 5 Jahre alt. Beachten Sie den unterschiedlichen Gesichtsausdruck, der das Fortschreiten der neurologischen Beeinträchtigung zeigt
Patienten mit Sanfilippo-Syndrom zeigen im zweiten und dritten Lebensjahr eine fortschreitende kognitive Beeinträchtigung. Im Gegensatz zur Mehrheit der MPS sind somatische Merkmale relativ weniger offensichtlich und die ZNS-Beteiligung ist auffällig. Die Verzögerung der Sprachentwicklung ist normalerweise das erste Anzeichen, das von den Eltern bemerkt wird, aber die Hauptprobleme können Verhaltensprobleme sein (Hyperaktivität, ängstliches und aggressives Verhalten, Schlafstörungen), gefolgt von fortschreitendem geistigem Verfall . Diese Symptome können Fehldiagnosen wie Verhaltensstörungen, Aufmerksamkeitsdefizit-Hyperaktivitätsstörung (ADHS) und Autismus-Spektrum-Störungen verursachen . Epilepsie kann ebenfalls vorhanden sein.
Schlafstörungen, deren Inzidenz bis zu 80-90% beträgt , sind ein häufiges Merkmal. Sie bestehen aus Schwierigkeiten beim Einschlafen und häufigem nächtlichen Erwachen mit vollständiger Umkehrung des Tag–Nacht-Rhythmus bei einigen Patienten.
All diese Manifestationen sind sehr schwer zu bewältigen und haben enorme psychologische Auswirkungen auf die Lebensqualität der ganzen Familie.
Obwohl der Phänotyp in den vier Subtypen sehr ähnlich sein kann, scheint der klinische Verlauf bei Sanfilippo B und C im Allgemeinen durch einen weniger schweren Phänotyp gekennzeichnet zu sein .
Nicht-neurologische Symptome sind bei MPS III meist weniger ausgeprägt als bei den anderen MPS. Wiederkehrende Hals-, Nasen- und Ohrenentzündungen werden in einem jüngeren Alter beobachtet. Wiederkehrende Mittelohrentzündungen, Mittelohrknöchelchendefekte und Anomalien im Innenohr können Taubheit verursachen. Ein Übermaß an dicken Sekreten und anatomischen Veränderungen kann zu einer Verstopfung der Atemwege führen. In den ersten Lebensjahren wird häufig über Durchfall berichtet, während Verstopfung bei älteren Patienten häufiger auftritt.
Die körperliche Untersuchung kann grobe Gesichtszüge zeigen, obwohl diese weniger ausgeprägt sind. Die Patienten haben Makrozephalie, breite Augenbrauen mit medialer Abfackelung und Synophrys, das Haar ist normalerweise trocken und grob und Hypertrichose ist häufig. Bei jüngeren Patienten werden häufig leichte Hepatomegalie sowie Nabel- und Leistenhernien beobachtet. Seltene und oft milde Kontrakturen finden sich meist in den Ellenbogen. Das Wachstum ist bei etwa der Hälfte der Patienten älter als 12 Jahre betroffen .
Zusammenfassend weist die MPS III- oder Sanfilippo-Krankheit eine ausgeprägte neurologische Beteiligung mit sehr milden somatischen Anzeichen auf. Kleinkinder im Alter von 2-3 Jahren, die Hyperaktivität, ADHS und aggressives Verhalten entwickeln, können im Verdacht stehen, MPS III zu haben.
MPS mit vorherrschender somatischer Beteiligung
MPS IV und MPS VI weisen nur eine somatische Beteiligung auf. Dies sind progressive Zustände, die die intellektuellen Fähigkeiten schonen und hauptsächlich das Skelett (MPS IVA) oder alle Organe / Systeme des Körpers (MPS VI) betreffen. Vererbung ist autosomal-rezessiv. Die Rate, mit der sich die Symptome verschlimmern, variiert zwischen den betroffenen Personen.
Mukopolysaccharidose IV (MPS IV, auch bekannt als Morquio-Syndrom; Abb. 8) stellt sich als zwei genetisch unterschiedliche Erkrankungen mit jeweils unterschiedlichem Enzymmangel dar: MPS IVA (Morquio A-Syndrom; OMIM # 253000) aufgrund eines Mangels an N-Acetylgalactosamin-6-sulfatase (GALNS-Gen), was zu einer Akkumulation von Keratansulfat (KS) und CS führt; und MPS IVB (Morquio B-Syndrom; OMIM # 253010) aufgrund eines Mangels an Beta-Galactosidase-Aktivität, die wie bei GM1-Patienten zu einer Ausscheidung von KS und Oligosacchariden im Urin führt. MPS IVB ist zwar allelisch für die verschiedenen Formen der GM1-Gangliosidose, es fehlt jedoch die psychomotorische Verschlechterung, die bei der GM1-Gangliosidose beobachtet wird .

MPS IVA. ein 4-jähriger Patient mit b antero-posterioren und c lateralen Röntgenaufnahmen, die “paddelförmige” Rippen und abgeflachte und abgerundete Wirbel mit einem typischen “vorderen Schnabel” -Aspekt zeigen
Patienten mit schwerer MPS IVA ohne nachweisbare Enzymaktivität zeigen ihre ersten Symptome in der Regel im ersten Lebensjahr, obwohl sie selten vor dem 4.-5. Lebensjahr diagnostiziert werden. Informationen zur Naturgeschichte von Morquio A. stammen hauptsächlich aus zwei umfangreichen Datensammlungen . Die reduzierte Wachstumsrate beginnt im Alter von ungefähr 18 Monaten und das Wachstum stoppt im Alter von ungefähr 7 oder 8 Jahren. Im Gegensatz zu den anderen MPS sind Morquio-A-Gelenke normalerweise locker und sehr flexibel (hypermobil), aber einige Gelenke (normalerweise Hüften und Schultern) können einen eingeschränkten Bewegungsbereich haben. Patienten mit einer solchen Skelettbeteiligung können eine Diagnose von Spondyloepiphysendysplasie, Pseudoachondroplasie, multipler Epiphysendysplasie oder bilateraler Legg-Calvé-Perthes-Krankheit erhalten oder untersucht werden .
Eine Konstante ist die Odontoidhypoplasie; so kann es zu einer Dislokation des Atlanto-Occipital-Gelenks kommen, die zu einer Kompression und Schädigung des Bulbärs und des Rückenmarks führt, was zu Lähmungen oder sogar zum Tod führt, wenn die zervikale Stabilisierung nicht frühzeitig durchgeführt wird.
MPS IVB-Patienten haben eine sehr ähnliche klinische Manifestation, jedoch mit einer weniger ausgeprägten Kleinwüchsigkeit. Sie haben leicht grobe Gesichtszüge, Hörverlust, Hornhauttrübungen, Herzklappenerkrankungen und häufige Infektionen der oberen Atemwege. Sie können auch mit Leistenbruch und leichter Hepatomegalie auftreten. Skelettdysplastische Merkmale umfassen Platyspondylie, Odontoidhypoplasie mit möglicher zervikaler Subluxation, Kypho-Skoliose, Coxa Valga und verengte Beckenflügel .
Mukopolysaccharidose VI (MPS VI, auch bekannt als Maroteaux-Lamy-Syndrom; OMIM #253200; Abb. 9) ist auf pathogene Mutationen im Arylsulfatase B (ARSB) -Gen zurückzuführen, das sich auf Chromosom 5 befindet. Infolgedessen beeinträchtigt eine verminderte oder fehlende Aktivität des Enzyms Arylsulfatase B (ASB) (N-Acetylgalactosamin-4-sulfatase) den Abbau von DS und seine zelluläre Akkumulation .

MPS VI. Ein 2-jähriger Junge mit offensichtlichem grobem Gesicht und Skelettdysostose
Patienten ohne nachweisbare Enzymaktivität haben die schwere Form von MPS VI und zeigen Symptome in der frühen Kindheit. Ihr somatischer Phänotyp ähnelt dem Hurler-Syndrom. In den meisten Fällen, im Alter von 2 oder 3 Jahren, werden schwere und fortschreitende Skelettschäden, Dysostose Multiplex genannt, offensichtlich. Diese Skelettdysplasie umfasst Hüftdysplasie mit dysplastischem Femurkopf, abnormalen Wirbelkörpern, unregelmäßigen Schlüsselbeinen, hypoplastischer distaler Ulna und Radius sowie dysplastischen und kurzen Mittelhandknochen, Handwurzel- und Fußwurzelknochen sind hypoplastisch und haben ein unregelmäßiges Profil. Die Wachstumsgeschwindigkeit verlangsamt sich häufig nach dem ersten Lebensjahr, wobei die Endhöhe im Allgemeinen unter 120 cm liegt. Wie in anderen Fällen ist ein frühes Zeichen grobe Gesichtszüge. Darüber hinaus haben Patienten einen typischen vorstehenden Bauch, Hepatomegalie, Nabel- und / oder Leistenbruch und Herzbeteiligung in der frühen Kindheit. Obwohl keine primäre geistige Behinderung vorliegt, kann bei einigen Patienten ein Hydrozephalus mit nachfolgender intrakranieller Hypertonie und Papillenödem beobachtet werden. Pectus carinatum, steife und kontrahierte Gelenke, Skoliose und Kyphose sowie die Kompression des zervikalen Rückenmarks verschlechtern sich mit der Zeit .
Zusammenfassend zeigen MPS IV und VI keine ZNS-Beteiligung. MPS IV ist ein einzigartiges MPS, das Gelenklaxheit zeigt, während der schwere Beginn von MPS VI dem bei MPS I.