Autosomalna recesywna wrodzona rybia łuska / Actas Dermo-Sifiliográficas
wprowadzenie
najnowsza klasyfikacja rybiej łuski rozróżnia 2 główne formy: Formy niesyndromiczne, które występują tylko z objawami skórnymi, oraz formy zespołowe, które występują również z objawami w innych narządach (Tabela 1).1 wśród form niesyndromicznych zidentyfikowano 4 grupy: wspólne ichtiozy, autosomalne recesywne wrodzone ichtiozy (ARCIs), keratynopatyczne ichtiozy i inne mniej powszechne ichtiozy.Tradycyjnie grupę ARCIs podzielono na 2 zaburzenia, lamellar ichthyosis (LI) i wrodzonej ichthyosiform erytrodermia (CIE). W nowej klasyfikacji do tej grupy dodano arlekinową rybią łuskę (HI) 1, ponieważ zidentyfikowano inaktywujące mutacje w genie ABCA12 jako odpowiedzialne za to zaburzenie,2, 3, podczas gdy nonsensowne mutacje w tym samym Genie mogą prowadzić do fenotypu LI4 lub CIE5, 6. Inne mniej powszechne warianty zawarte w grupie ARCIs są self-healing collodion baby (SHCB), acral SHCB i rybia łuska kostium kąpielowy.7-9
Klasyfikacja Konsensusowa oparta na cechach klinicznych Ichtiozy1.
| Недромальные formy | Синдромные formy |
| Popularnym ихтиоз, wulgarne ихтиоз, recesywny x-сцепленный ихтиоз (несиндромный ), podstawowe formy шарлекинового ихтиоза, płytkowy ихтиоз, wrodzony ихтиосиформный эритродермит lub formy самовосстанавливающегося коллодиона, dzieci w krzyżowy коллодион, strój do kąpieli, ихтиоз, кератинопатический ихтиоз, podstawowe formy эпидермолитического ихтиоза, powierzchowny эпидермолитический ихтиоз drobne формыаннулярный эпидермолитический ихтиозискурт-ихтиоз Mclean autosomalny recesywny эпидермолитический ихтиозэпидермолитический невусдругие formy хлорикриновой кератодермии, эритрокератодермия, zmienność skóry, zespół, zespół конгенитальной siatkowej ихтиосиформной zapalnych, zespół Kliknięcia | Zespół X-połączonego ихтиосрецессивного x-połączonego ихтиоза (zespół), фолликулярный ихтиоз, łysienie i światłowstręt (IFAP) zespół конради-Скамманна-хаппла ( punktowy хондродисплазия 2 rodzaje)zespół аутосомного ихтиоза skóry zaburzenia zespół Нетертона zespół ихтиоза-гипотрихоза zespół ихтиоза-stwardniające zapalenie dróg żółciowych, zespół ихтиострофиневрологические zaburzenia Sjögrena-Larssona syndromeRefsum diseaseMEDNIK syndromeFatal disease courseGaucher disease, type 2Multiple sulfatase deficiencyCEDNIK syndromeARC syndromeOther associated signsKID syndromeChanarin-Dorfman syndromeIchthyosis prematurity syndrome |
Abbreviations: ARC, arthrogryposis–renal dysfunction–cholestasis; ARCI, autosomal recessive congenital ichthyosis; CEDNIK, cerebral dysgenesis, neuropathy, ichthyosis, and palmoplantar keratoderma; KID, keratitis ichthyosis deafness; KLICK, keratosis linearis with ichthyosis congenital and sclerosing keratoderma; MEDNIK, upośledzenie umysłowe, enteropatia, głuchota, neuropatia obwodowa, rybia łuska, rogowacenie skóry.
dostępne są jedynie ograniczone dane dotyczące epidemiologii ARCIs. W Stanach Zjednoczonych szacuje się, że częstość występowania przy urodzeniu wynosi 1 na 100 000 populacji w przypadku LI i 1 na 200 000 populacji w przypadku CIE. Inne badania donoszą o łącznym występowaniu LI I CIE od 1 na 200 000 do 300 000 populacji.10,11 w niektórych krajach, takich jak Norwegia, szacowana częstość występowania jest większa (1 Na 91 000) ze względu na mutacje założycielskie.12 odkrycie 1 lub kilku powtarzających się mutacji w populacji może być spowodowane tym, że mutacja wystąpiła w danym momencie historii i była następnie przekazywana z pokolenia na pokolenie (mutacja założycielska) lub dlatego, że region genomu, w którym znajduje się mutacja, ma sekwencję DNA podatną na mutację (mutacja hotspot). W Hiszpanii szacowana częstość występowania ARCI wynosi 1 na 138 000 w populacji ogólnej i 1 na 61 700 wśród dzieci poniżej 10 roku życia.13 w niektórych regionach Hiszpanii częstość występowania może być jeszcze wyższa. Na przykład na wybrzeżu Galicji odnotowano występowanie 1 na 33 000, również ze względu na efekt założycielski.
płytkowa rybia łuska i wrodzona rybia łuska cechy Erytrodermakliniczne
chociaż początkowo uważano, że LI I CIE są różnymi jednostkami, istnieją doniesienia o pacjentach z pośrednimi objawami klinicznymi i oba warunki mogą być spowodowane mutacjami w tym samym Genie.15,16 ponadto u pacjentów z tą samą mutacją, nawet w obrębie tej samej rodziny, mogą rozwinąć się różne fenotypy.12,15
większość pacjentów rodzi się otoczona błoną kolodionową, która stopniowo zanika w pierwszych tygodniach życia i zostaje zastąpiona definitywnym fenotypem (Fig. 1a). Hipohydroza, ciężka nietolerancja ciepła i dystrofia paznokci są często obserwowane zarówno w LI, jak i CIE.U 17-19 pacjentów z LI zwykle występują cięższe objawy kliniczne niż u pacjentów z CIE. Mają duże, płytkowate łuski, często ciemnego koloru, pokrywające całą powierzchnię ciała. Erytrodermia jest nieobecny lub minimalne. Tacy pacjenci zwykle mają ektropion, a czasami eclabium, niedorozwój chrząstki stawowej i nosowej, bliznowate łysienie, zwłaszcza na krawędzi skóry głowy i keratodermy palmoplantar (Fig. 1B i C). CIE charakteryzuje się obecnością erytrodermy i drobnego białawego skalowania (Fig. 2). U niektórych pacjentów zaznaczono rumień i uogólnione skalowanie. Łuski mogą być duże i ciemne, szczególnie na powierzchniach prostowników nóg. W mniej ciężkich przypadkach rumień jest łagodny, a skalowanie jest w porządku.

cechy kliniczne rybiej łuski blaszkowej . A, brązowawo-blaszkowe złuszczanie. B, zaznaczona hiperkeratoza podeszwowa. C, bliznowate łysienie skóry głowy.
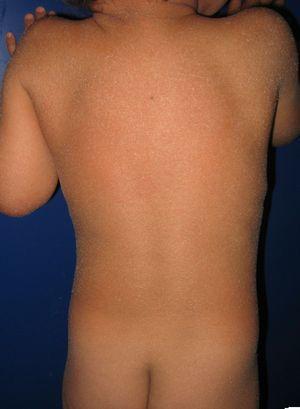
pacjent z wrodzoną erytrodermą ichtiozoiczną i mutacjami w genie ALOXE3. Można zauważyć łagodny rumień i uogólnione białawe złuszczanie futerkowe.
histopatologia
zmiany histopatologiczne nie dają rozpoznania. U LI obserwuje się masywną hiperkeratozę ortokeratotyczną, zwykle z dwukrotnym wydłużeniem jak u CIE. Naskórek jest akantotyczny i czasami przybiera wygląd podobny do łuszczycy. Tempo proliferacji komórek jest normalne lub nieznacznie podwyższone.17-19 pacjenci z CIE mają mniej zaznaczoną hiperkeratozę, z ogniskową lub rozległą parakeratozą, normalną lub zagęszczoną warstwą ziarnistą i bardziej wyraźną akantozą. Zwiększa się rotacja naskórka.17-19
Ultrastruktura
chociaż do tej pory nie znaleziono ścisłej korelacji między wynikami molekularnymi, klinicznymi i ultrastrukturalnymi, mikroskopia elektronowa może być jednak przydatna do wykluczenia innych form rybiej łuski i do prowadzenia analiz genetycznych w niektórych przypadkach. Opisano cztery rodzaje wrodzonej rybiej łuski (Tabela 2).
Klasyfikacja Ultrastrukturalna wrodzonych Ichtioz.
| Typ | główna cecha | pozostałe cechy | mutacje | objawy kliniczne |
| 1 | nieobecność markerów ultrastrukturalnych rybiej łuski typu 2, 3 i 4 | kropelki lipidowe lub pierścienie w warstwie rogowej (najczęściej) małe granulki keratohyaliny grudki pęcherzykowe lub płatkowe granulki powłoki membranowej | tgm1 (33 .3%) ALOX12B (2 przypadki) | CIE |
| 2 | rozszczepy cholesterolu w warstwie rogowej | brak lub przerzedzenie otoczek rogowychmałe granulki keratohjaliny krople lipidu | TGM1 (89-100%) | LI |
| 3 | laminowane struktury błonowe w warstwie granulosum i / lub warstwy rogowej. | nieprawidłowe granulki powłoki membranowej krople lipidowefoci wybitnych wakuoli juxtanuclear w warstwie ziarnistej | NIPAL4 (93%) | CIE (najczęściej)LI |
| 4 | Trilamellar membrane packets that fill some cells in the stratum granulosum and / or stratum corneum | anormal membrane coating granules | FTAP4 | Ichthyosis prematurity syndrome (100%) |
skróty: CIE, wrodzona rybia łuska erytrodermia; LI, rybia łuska płytkowa.
wrodzona rybia łuska typu 1
wrodzona rybia łuska typu 1 charakteryzuje się brakiem markerów ultrastrukturalnych dla rybiej łuski typu 2, 3 i 4. Dlatego diagnoza jest zwykle tylko wtedy, gdy inne typy zostały wykluczone. Najczęstszym stwierdzeniem jest obecność kropel lub pierścieni lipidowych w warstwie rogowej (rys. 3A).20 te krople lipidowe nie są stałą cechą lub specyficzne dla tego konkretnego typu, ponieważ nie są obecne we wszystkich przypadkach, 20 i mogą być obecne w innych typach rybiej łuski.21,22 klinicznie u większości pacjentów występują objawy CIE.12,20 jedna trzecia pacjentów ma mutacje w genie TGM1.Ten typ ultrastrukturalny został również zidentyfikowany w związku z mutacjami w genie ALOX12B.23,24

obrazy z mikroskopu elektronowego. A, wrodzona rybia łuska typu 1, pokazując kropelki lipidów w warstwie rogowej i brak markerów ultrastrukturalnych innych rodzajów rybiej łuski. B, wrodzona rybia łuska typu 2, charakteryzuje się obecnością cholesterolu rozszczepy (strzałka) w korneocytach.
wrodzona rybia łuska typu 2
wrodzona rybia łuska typu 2 charakteryzuje się rozpadami cholesterolu w warstwie rogowej (rys. 3b).21 takie rozszczepy są stałym stwierdzeniem w tego typu rybiej łuski i mogą być wykryte w różnych biopsjach u tego samego pacjenta; leczenie retinoidami doustnymi nie ma wpływu na te rozszczepy.U niektórych pacjentów z niedoborem aktywności Tgazy 1 zaobserwowano również 12,25 agregatów gęstych elektronowo na korneocytach.26-28 klinicznie, u większości pacjentów występują ciężkie objawy CIE.Ten ultrastrukturalny typ jest silnie związany z mutacjami w genie TGM1.12,16
wrodzona rybia łuska typu 3
wrodzona rybia łuska typu 3 charakteryzuje się błoniastymi strukturami w warstwie ziarnistej i / lub warstwie rogowej. Struktury te są rozmieszczone w paski wokół pustej przestrzeni blisko jądra.22,29-31 objawy kliniczne tego typu różnią się od innych; początek rybiej łuski jest zmienny, złuszczanie i rumień może być niejednolity lub uogólniony, a zginanie w szczególności ma wpływ. Mutacje w genie NIPAL4 odpowiadają za 93% ichtioz typu 3.32
wrodzona rybia łuska typu 4
charakterystyczne jest, że w wrodzonej rybiej łusce typu 4, niektóre komórki warstwy ziarnistej i warstwy rogowej są wypełnione pakietami błon trójgwiazdowych.33 te odkrycia są patognomic dla rybiej łuski zespół wcześniaków, stan obecnie uważany za formę zespołu rybiej łuski.34,35
badania molekularne
pod względem genetycznym ARCIs są bardzo niejednorodne. Gen TGM1 jest związany z większością przypadków, ale odnotowano mutacje w 5 innych genach (ALOX12B, ALOXE3, NIPAL4, CYP4F22 i ABCA12). Fischer i in.36 zbadało 520 rodzin z ARCI i zidentyfikowało mutacje w co najmniej 1 z tych genów w 78% przypadków (TGM1 w 32%, NIPAL4 w 16%, ALOX12B w 12%, CYP4F22 w 8%, ALOXE3 w 5% i ABCA12 w 5%). W innym badaniu z udziałem 250 pacjentów z ARCI o różnym pochodzeniu, 38% miało mutacje TGM1, 6,8% miało mutacje ALOXE3, a 6,8% miało mutacje ALOX12B.37 w Galicji zidentyfikowaliśmy mutacje w genach TGM1, ALOX12B, ALOXE3, NIPAL4 i CYP4F22 w 75% badanych rodzin, ale rozmieszczenie mutacji było inne.14 Gen TGM1 zmutowano w 68.7% przypadków, podczas gdy gen ALOXE3 został zmutowany tylko u 1 pacjenta. Nie wykryliśmy mutacji w żadnym z trzech badanych genów.
TGM1
Gen TGM1 znajduje się na chromosomie 14q11.2 i ma 15 eksonów (GenBank NM-000359.2). Koduje enzym Tgazy 1, który jest jednym z 3 enzymów tgazy występujących w naskórku.Enzym ten uczestniczy w tworzeniu zakrzywionej otoczki poprzez katalizowanie sieciowania kilku białek zależnych od wapnia, takich jak involukryna, lorikryna i białka bogate w prolinę.39,40 katalizuje również Wiązanie ??- hydroksyceramidy w zewnętrznej warstwie zakrzywionej koperty z białkami w wewnętrznej warstwie.41,42 u pacjentów z mutacjami TGM1 brakuje zrogowaciałej otoczki i aktywność Tgazy 1 jest zmniejszona lub nie istnieje.43-47
od 1995 r., kiedy gen ten został zidentyfikowany jako odpowiedzialny za niektóre przypadki ARCI, u pacjentów o różnym pochodzeniu odnotowano 48-50 więcej niż 110 mutacji. Mutacje w TGM1 są najczęstszą przyczyną ARCI.36,37 mutację tę stwierdzono w 55% przypadków w Stanach Zjednoczonych i w 84% przypadków w Norwegii.12,51 najczęstszą mutacją jest ok.877-2A>g, co stwierdzono u 34% zmutowanych alleli zgłoszonych do tej pory.52 wysoka częstotliwość występowania tej mutacji w krajach takich jak Stany Zjednoczone i Norwegia wynika z efektu założycielskiego.12,53 drugą najczęstszą mutacją jest P. Arg142His. Ta i podobne mutacje zostały zgłoszone w krajach takich jak Egipt, Niemcy, Finlandia i Stany Zjednoczone, 15,49-51,54-56 i wydaje się,że są to mutacje hotspot.Mutacja P. Arg307Trp jest częsta w populacji japońskiej.5 w Galicji, P. Arg760X, c. 1223_1227delACACA i c.984 + 1G>a mutacje w TGM1 zidentyfikowano w 81,82% rodzin z mutacjami w tym genie, co sugeruje efekt założycielski.14 potwierdzeniem tej hipotezy było badanie haplotypu (praca dotychczas niepublikowana).
mutacje TGM1 są odpowiedzialne za większość przypadków LI15,27,44,46,56,58-63 i dla niewielkiego odsetka przypadków CIE.43,47,64,65 takie mutacje mogą również powodować inne formy ARCI, takie jak SHCB, acral SHCB i rybia łuska kostiumu kąpielowego.
w wielu badaniach próbowano wykazać związki genotypowo-fenotypowe pomiędzy mutacjami w TGM1 i ultrastrukturalnymi lub klinicznymi, ale do tej pory nie zaobserwowano znaczącej korelacji.15,16,53 ogólnie rzecz biorąc, pacjenci z mutacjami w genie TGM1 są bardziej dotknięci niż ci bez takich mutacji. W badaniu z udziałem 83 pacjentów z ARCI w Szwecji i Estonii obecność ektropionu i kolodionu baby była związana z mutacjami TGM1, podczas gdy wyższy wskaźnik rumienia obserwowano u pacjentów bez mutacji w tym genie.Inne badanie wykazało, że rodzaj skalowania jest główną różnicą między nosicielami i osobami niebędącymi nosicielami mutacji TGM1, stwierdzając, że wszyscy pacjenci z mutacjami w tym genie mieli skalowanie blaszkowe, podczas gdy 80% pacjentów bez mutacji tgm1 miało skalowanie drobne.Ponadto zaobserwowano, że mutacje skracające są częściej związane z zaburzeniami hipohydrozy i pocenia się niż mutacje typu missense.51 w populacji północnoamerykańskiej model oparty na obecności pewnych cech klinicznych przewiduje, że u pacjentów urodzonych jako dzieci z kolodionem i z zaburzeniami ocznymi i/lub łysieniem występuje 4 razy większe prawdopodobieństwo wystąpienia mutacji TGM1.51
aloxe3 i ALOX12B
geny ALOXE3 i ALOX12B znajdują się na chromosomie 17p13.1.67 mają podobną strukturę z 15 eksonami kodującymi naskórek loxs eLOX-3 i 12R-LOX.68,69 fakt, że wyrażają się one głównie w ponadpodstawowych warstwach naskórka, wspiera ich rolę w zaawansowanych fazach różnicowania się naskórka, z udziałem w przetwarzaniu ciał blaszkowych.24,70 enzymy te działają na sąsiednie etapy szlaku hepoksylinowego (Fig. 4). 12R-LOX przekształca kwas arachidonowy w kwas 12R-hydroksyeikosatetraenowy, podczas gdy eLOX – 3 przekształca ten produkt w izomer69,71 epoksyalkoholu z rodziny hepoksylin A3.Produkt hepoksyliny jest niestabilny i hydrolizowany w komórkach do specyficznej pochodnej trihydroksyliny (trioksyliny). Chociaż dokładna rola produktów szlaku hepoksylinowego nie jest znana, spekulowano, że mogą one uczestniczyć w tworzeniu międzykomórkowych lipidów warstwy rogowej lub działać jako sygnały indukujące różnicowanie keratynocytów.
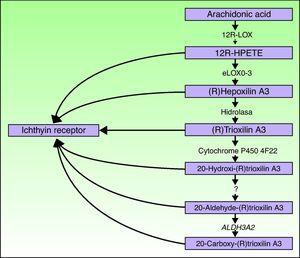
schemat szlaku hepoksylinowego, pokazujący udział genów ALOXE3, ALOX12B, NIPAL4 i CYP4F22. Mutacje w tych genach są odpowiedzialne za niektóre rodzaje ARCI. HPETE wskazuje na kwas hydroperoxyeicosatetraenoic.
geny ALOX12B i ALOXE3 zidentyfikowano po raz pierwszy w 2002 r. 73,74 od tego czasu odnotowano ponad 30 mutacji w genach ALOX12B23, 24,37,75-77 i około 10 w genach ALOXE337,74,75. Mutacje te odpowiadają za 14% do 17% ARCIs36,37 i 72.2% SHCBs.23,78,79 związek przyczynowy między tymi mutacjami a fenotypem został potwierdzony poprzez wykazanie,że aktywność katalityczna LOX naskórka została całkowicie zniesiona u pacjentów z tymi mutacjami75, 80 oraz poprzez wykorzystanie modeli zwierzęcych, które odtworzyły fenotyp ichtiozoiczny u ludzi.81-83 oba geny odpowiadają za podobny odsetek przypadków ARCI. Jednak zakres różnych mutacji w genie ALOXE3 jest ograniczony, ze względu na przewagę 2 mutacji, p. Arg234X i P. Pro630Leu, które wydają się odpowiadać hotspotom.37, 74, 75
pacjenci z mutacjami w genach ALOXE3 i ALOX12B zwykle wykazują fenotyp CIE.74,75,77 nasilenie skalowania jest łagodne lub umiarkowane, a łuski mają białawy lub jasnobrązowy kolor. Rumień może być również obecny. Aż 76% pacjentów rodzi się jako niemowlęta kolodionowe, a 88% ma zaburzenia pocenia się.U 37 pacjentów z mutacjami w genie ALOX12B występuje bardziej ograniczone, białawe złuszczanie w porównaniu z nosicielami mutacji w genie ALOXE3. W takich przypadkach łuski są brązowawe i przylegające. Obecność rumienia, hiperkeratozy dłoni i podkreślenia fałdów dłoni są również związane z mutacjami ALOX12B.37
Ichthyin/NIPAL4
Gen NIPAL4, znany również jako gen ICHTHYIN, znajduje się na chromosomie 5q33. Ma 6 eksonów kodujących białko z kilkoma domenami transbłonowymi o nieznanej funkcji.Postawiono hipotezę, że produkt białkowy uczestniczy w tym samym szlaku metabolicznym co LOX i może działać jako receptor dla trioksylin A3 i B3 lub dla innych metabolitów szlaku metabolicznego hepoksyliny.84 miałoby to zatem związek z formowaniem ciał blaszkowych lub ich transportem w kierunku przestrzeni zewnątrzkomórkowej.32 na poparcie tego są 2 obserwacje. Po pierwsze, w 93% przypadków, mutacje w tym genie są związane z ultrastrukturalny wzór wrodzonej rybiej łuski typu 3, charakteryzuje się nieprawidłowościami w ciałach blaszkowych i obecności wydłużonych błon okołopądkowych w warstwie granulosum.32 Po Drugie, NIPAL4 ulega ekspresji zasadniczo w warstwie ziarnistej naskórka, gdzie obecne są ciała blaszkowe.85
od odkrycia genu NIPAL4 w 2004 r. 84 odnotowano tylko 9 mutacji u pacjentów z krajów śródziemnomorskich (Algieria, Turcja i Syria), 84 krajów skandynawskich,32 Pakistanu,85 Wysp Owczych, 32 i Ameryki Południowej.
spektrum kliniczne pacjentów z mutacjami w tym genie jest szerokie, nawet wśród członków tej samej rodziny. Od 3,7%32 do 60%84 rodzą się jako Dzieci kolodionowe. Gdy błona kolodionowa znika, u większości pacjentów rozwijają się objawy CIE, z drobnymi białawymi łuskami na rumieniowej podstawie na twarzy i tułowiu oraz większymi, brązowawymi Łuskami na szyi, pośladkach i nogach.Mogą występować wyraźne kseroza, uogólnione brązowawe siateczkowe płytki hiperkeratotyczne, które wydają się zaakcentowane w fałdach skórnych oraz dyschromia twarzy.32,85 ponadto, keratoderma palmoplantar jest częstym stwierdzeniem wraz z okazjonalnymi przykurczami palców i zakrzywionymi paznokciami. Niektóre badania donoszą o bardziej typowych dla LI odkryciach.U niektórych pacjentów zgłaszano występowanie objawów przedmiotowych i podmiotowych atopowego zapalenia skóry, chociaż w żadnym z tych przypadków nie wykryto mutacji w genie FLG.
CYP4F22
Gen FLJ39501 lub CYP4F22 znajduje się na chromosomie 19p13.12.86 ma 12 egzonów87 i koduje cytochrom P450, rodzinę 4, podrodzinę F, polipeptyd 2, homolog leukotrienu B4 – ω-hydroksylazy (CYP4F2). Reakcję katalizowaną przez produkt flj39501 w skórze i substratach tej reakcji można wywnioskować przez analogię z jej znanymi homologami CYP4F2 i CYP4F3.Postawiono hipotezę, że CYP4F2 i CYP4F3 uczestniczą w szlaku heptoksylinowym poprzez katalizowanie konwersji trioksyliny A3 do 20-hydroksy – (r)trioksyliny A387 i że produkt końcowy tego szlaku, 20-karboksy-trioksylina A3, może mieć kluczowe biologiczne działanie regulacyjne w skórze.89
do tej pory odnotowano tylko 8 mutacji tego genu w 12 spokrewnionych rodzinach z krajów Śródziemnomorskich87 i w 1 rodzinie pochodzenia Izraelskiego.62
w rodzinach zgłoszonych przez Lefèvre et al., 87 większość pacjentów miała fenotyp CIE przy urodzeniu i ten następnie postępował do LI. pacjenci urodzili się zwykle z zaznaczoną erytrodermą, chociaż bez błony kolodionowej. Wraz z wiekiem wykształciły się u nich uogólnione białawo-szare skalowanie, które było bardziej zaznaczone w okolicy okołomięśniowej, na pośladkach i dolnej części ciała. Hiperlinearność dłoni i podeszew oraz złuszczanie na skórze głowy, w czasach typu pityriasiform, były częste.87 w innej rodzinie, 3 członków dotkniętych urodziło się jako Dzieci collidion i rozwinęła intensywną erytrodermę, uogólnione złuszczanie i keratodermę palmoplantar.62
ABCA12
w 2003 r. stwierdzono, że Gen ABCA12 jest odpowiedzialny za niektóre przypadki LI i został zmapowany na chromosomie 2q34. 4. Następnie potwierdzono, że mutacje w tym genie są również odpowiedzialne za HI.2, 3ABCA12 koduje 53 eksony i należy do rodziny transporterów ABC, które wiążą adenozynotrójfosforan, jednocześnie ułatwiając transport kilku cząsteczek przez błonę komórkową.90 członkowie podrodziny ABCA są zaangażowani w transport lipidów.Niedobór funkcji ABCA12 powoduje zaburzenia transportu lipidów w ciałach blaszkowych, co prowadzi do zmniejszenia międzykomórkowego poziomu lipidów w warstwie rogowej.Badania ultrastrukturalne wykazały, że ABCA12 znajduje się w ciałach blaszkowych związanych z glikozyloceramidami.Mutacje 91ABCA12 były związane z zaburzeniami w dystrybucji i transporcie glikozyloceramidów oraz ze zmniejszonym poziomem hydroksyceramidów, jednego z głównych składników bariery lipidowej w przestrzeniach międzykomórkowych.3,6,92,93 masywna hiperkeratoza występująca u tych pacjentów może być odpowiedzią kompensacyjną na niedobór bariery lipidowej.Może to być również spowodowane brakiem złuszczania korneocytów, 93 co może być spowodowane przez defekty w transporcie niektórych proteaz, takich jak kallikreina 5 i katepsyna D, wynikające z zaburzeń w ciałach blaszkowych.Modele Mysie i badania in vitro sugerują, że mutacje ABCA12 mają również wpływ na różnicowanie naskórka.95-97
do tej pory odnotowano ponad 50 mutacji w genie ABCA12 u pacjentów z ARCI z Afryki, Europy, Pakistanu i Japonii. Najczęstsze mutacje to P. Val244SerfsTer28,2,98,99 zidentyfikowane w populacjach pakistańskich i indyjskich oraz P. Asn1380Ser, 4 zidentyfikowane w rodzinach afrykańskich. W obu przypadkach mogą to być mutacje założycielskie.
zakres mutacji ABCA12 jest związany z fenotypem, z mutacjami związanymi z całkowitą utratą funkcji prowadzącą do fenotypu HI.2,3,98-102 natomiast w LI I CIE większość mutacji ma charakter missense i ma mniej poważny wpływ na funkcję białka.4-6, 103 mutacje leżące u podstaw fenotypu LI wydają się być skoncentrowane w pierwszym regionie kasety wiążącej adenozynotrójfosforan.Klinicznie u pacjentów z CIE i mutacjami w genie ABCA12 skala średniej wielkości jest nieco większa niż u pacjentów z tym fenotypem.
rybia łuska Arlekina
HI lub płód Arlekina jest ciężką i zwykle śmiertelną formą rybiej łuski. Dzieci są zwykle przedwczesne z rozległymi błyszczącymi blaszkami hiperkeratotycznymi, oddzielonymi głębokimi szczelinami, które pokrywają całą powłokę i tworzą geometryczne wzory przypominające ubranie noszone przez arlekiny, dając tym samym warunek jego nazwę. Napięcie skóry prowadzi do wyraźnego zwinięcia powiek i warg, szczątkowego rozwoju chrząstki stawowej i nosowej oraz, czasami, małogłowia. Dzieci rzadko mają rzęsy lub brwi, chociaż włosy na skórze głowy mogą być zachowane. Dłonie i stopy są opuchnięte i obrzęknięte, często pokryte warstwą przypominającą rękawicę. Mogą mieć przykurcze palców.
w przypadku takich pacjentów ryzyko zgonu w okresie noworodkowym jest bardzo wysokie.104 wentylacja płucna jest zagrożona; przezepidermalna utrata wody prowadzi do odwodnienia, nierównowagi hydroelektrycznej i niestabilności termicznej; zwiększa się ryzyko infekcji. Ucisk twarzy i eclabium utrudniają ssanie, a tym samym karmienie, z odpowiednim pogorszeniem odwodnienia. Noworodki z tą chorobą rzadko żyły dłużej o kilka tygodni. Jednak w ostatnich latach szanse na długoterminowe przeżycie znacznie wzrosły, głównie dzięki podawaniu retinoidów układowych i postępowi w intensywnej opiece nad noworodkiem.W ostatnim badaniu przeżyło 83% pacjentów leczonych retinoidami doustnymi w porównaniu do 24% pacjentów nieleczonych. Większość zgonów nastąpiła w pierwszych 3 dniach życia, ale leczenie rozpoczęto dopiero po tym u wielu ocalałych.Sugeruje to, że wiele z tych wczesnych zgonów miało miejsce niezależnie od leczenia retinoidami.
u dzieci, które przeżyły okres noworodkowy, zwykle rozwija się ciężka CIE.Charakter i lokalizacja mutacji w genie ABCA12 oraz stopień utraty funkcji transportera mogą decydować o rokowaniu.3,92,107 pacjenci, którzy zachowują pewien stopień aktywności białek, choć są minimalni, mogą mieć większe szanse na przeżycie. Nosiciele mutacji homozygotycznych mają wyższy wskaźnik śmiertelności.104
główną cechą histologiczną HI jest obecność niezwykle grubej i zwartej warstwy ortokeratotycznej rogówki. Mieszki włosowe i przewody potowe mają widoczne zatyczki hiperkeratotyczne107, 108 i mają nieprawidłowe lub nieobecne organy blaszkowe, inkluzje lipidowe lub pozostałości organelli lub jąder w korneocytach oraz brak lipidów międzykomórkowych w badaniu ultrastrukturalnym.108,109 mieszki włosowe wykazują wyraźne stężenie materiału rogowacenia, co jest cechą diagnostyczną HI stosowaną w diagnostyce prenatalnej.
do tej pory szybkość wykrywania mutacji w genie ABCA12 u pacjentów z HI jest bliska 100%, a więc wydaje się, że jest to genetycznie jednorodna choroba.
collodion Baby I Self-healing collodion Baby
collodion dzieci zwykle rodzą się przedwcześnie, a zachorowalność i śmiertelność okołoporodowa są zwiększone. Po urodzeniu noworodek jest pokryty błyszczącą, przezroczystą błoną przypominającą owijanie celofanem (rys. 5). Dzieci mają ektropion, eclabium i hipoplazję chrząstki nosowej i stawowej. Może to utrudnić ssanie i wentylację płucną110 oraz przezepidermalną utratę wody i zwiększa się ryzyko zakażeń.110,111

dziecko Kolodionowe, które następnie rozwinęło się w fenotyp rybiej łuski.
Collodion baby to zwykła prezentacja dla HI i CIE. Autosomalnie dominujący LI,112,113 zespół Sjögrena-Larssona,110 trichotyodystrofia,114 młodzieńcza choroba Gauchera, 110 neutralna choroba przechowywania lipidów, zespół Conradiego-Hünermanna-Happle ‘ a, zespół Hays-Wellsa i dysplazja ektodermalna115 mogą również okazjonalnie występować jako dziecko kolodionowe. Błona zanika samoistnie u 10% do 24% noworodków, ustępując całkowicie normalnej skórze.110,116 w przeszłości przypadki te były opisywane jako LI noworodka, 117, ale nie są one określane jako SHCB.118 niektórzy autorzy zasugerowali termin samodoskonalenia rybia łuska kolodionowa, ponieważ wielu z tych pacjentów, po ponownym zbadaniu w późniejszym okresie dzieciństwa lub jako dorośli, ma zmienny stopień anhydrozy i nietolerancji ciepła oraz łagodne objawy rybiej łuski, takie jak kseroza i drobne złuszczanie, szczególnie w pachach i szyi.78
ani mikroskopia optyczna, ani badania ultrastrukturalne kolodionu dziecka nie są specyficzne. Dlatego lepiej jest opóźnić biopsję skóry, aż do uzyskania ostatecznego fenotypu.
mutacje w genach TGM1,7,119ALOXE3,78 i ALOX12B23,78,79 zostały zidentyfikowane u pacjentów z SHCB. Mutacje ALOX12B są najczęstsze. W serii 15 skandynawskich pacjentów z SHCB, 67% miało mutacje w genie ALOX12B, 25% w genie ALOXE3 i 8,3% w genie TGM1.78 mutacji nie stwierdzono u niektórych pacjentów, więc inne geny mogą być również powiązane. Spekulowano, że mutacje te zmniejszają aktywność enzymatyczną w macicy, ale nie po urodzeniu.7 w macicy, gdzie ciśnienie hydrostatyczne jest wysokie, chelatacja przez wodę przekształca zmutowany enzym w nieaktywną konformację. Po urodzeniu, gdy ciśnienie spada, enzym powraca do swojej aktywnej formy, a jego aktywność wzrasta wystarczająco, aby utrzymać normalny lub minimalnie zmieniony fenotyp.7
Acral Self-healing collodion Baby
chociaż collodion dziecko wpływa na całe ciało, przypadki ograniczone do obszarów akral zostały zgłoszone. W 1952, Finlay et al.120 zgłosiło przypadek błony kolodionowej, która miała wpływ tylko na dłonie i stopy, a następnie na samouzdrawiający się kurs. Niedawno odnotowano nowy przypadek acral SHCB w związku z mutacjami genu TGM1.Nie wiadomo, dlaczego zmiany te są ograniczone do obszarów akralnych, chociaż mogą działać czynniki związane z zależną od miejsca regulacją aktywności enzymów.8
rybia łuska w stroju kąpielowym
rybia łuska w stroju kąpielowym została po raz pierwszy zgłoszona jako niezależny wariant ARCI w 2005 r., chociaż wcześniej zgłaszano przypadki rybiej łuski o szczególnym rozkładzie.121-123 wykryto go głównie u pacjentów pochodzenia południowoafrykańskiego, 9 chociaż odnotowano go również u osób z Europy i krajów śródziemnomorskich.124 po urodzeniu pacjenci mają uogólnioną błonę kolodionową, która następnie opuszcza charakterystyczny rozkład skalowania. Tułów, proksymalny obszar ramion, w tym pachowe, szyja i skóra głowy są ogólnie dotknięte, podczas gdy centralna część twarzy, kończyny i region nadnerczy są zwykle oszczędzane.9 łuski są duże, blaszkowate i ciemne. Drobniejsze złuszczanie może wystąpić w Fossae podkolanowym i przedkubitowym.124,125 dłonie i podeszwy stóp mają łagodną rozproszoną hiperkeratozę, podczas gdy plecy dłoni i stóp nie wykazują żadnego zaangażowania.
badanie histopatologiczne skóry dotkniętej chorobą wykazało wyraźną hiperkeratozę bez parakeratozy, normalne warstwy ziarniste, łagodną lub umiarkowaną rogowacenie i łagodne nacieki limfatyczne w górnej skórze właściwej.9 obserwacje Mikroskopii Elektronowej są zgodne z wrodzoną rybią łuskę typu 2 w większości przypadków. Nierozpuszczona skóra nie wykazuje żadnych nieprawidłowości.124,125 w zdrowej skórze aktywność Tgazy 1 jest nieznacznie zmniejszona i zwykle zlokalizowana w obszarach perykomórkowych. W zaangażowanej skórze aktywność enzymatyczna jest szczątkowa i nieprawidłowo zlokalizowana w cytoplazmie.124
wykryto mutacje w genie TGM1 u wszystkich pacjentów z rybia łuska kostiumu kąpielowego badanych do tej pory.119,124-126 najczęstszą mutacją jest P. Arg315Leu, który został zidentyfikowany u większości pacjentów z RPA i może być mutacją założycielską. Oji i in.124 sugerował, że temperatura skóry może odgrywać rolę w rozwoju tych objawów. Wykorzystując termografię cyfrową, autorzy wykazali silną korelację między temperaturą ciała a złuszczaniem, przy czym najbardziej dotknięte są najgorętsze obszary ciała. Aufenvenne et al.127 wykazało spadek optymalnej temperatury dla aktywności Tgazy 1 u pacjentów z rybią łuską kostiumu kąpielowego. Tego spadku nie obserwowano u zdrowych kontrolerów ani u pacjentów z uogólnionym LI. ten spadek temperatury wyjaśniałby fenotyp tych pacjentów. Optymalna temperatura wynosi 37°C dla normalnego enzymu, ale 31 ° C dla zmutowanego enzymu.
leczenie
podstawowym celem leczenia rybiej łuski jest wyeliminowanie skalowania i zmniejszenie kserozy bez powodowania nadmiernego podrażnienia(Tabela 3). Przed podjęciem decyzji o leczeniu należy wziąć pod uwagę takie aspekty, jak wiek i płeć pacjenta, rodzaj i nasilenie choroby oraz zakres i miejsce zmian.128
Strategia terapeutyczna w autosomalnych recesywnych wrodzonych Ichtiozach.
| Strategia terapeutyczna autosomalnej recesywnej wrodzonej ichtiozy | |
| Kąpiel i mechaniczna eliminacja łusek | kąpiel wodorowęglanem sodu lub skrobią pszenną, skrobią kukurydzianą lub skrobią ryżową; mechaniczne usuwanie wagi (1 lub 2 razy dziennie) |
| leczenie miejscowe (sekwencyjne) | środki nawilżające zawierające mocznik leki nawilżające z glikolem propylenowym połączone środki keratynolityczne (glikol propylenowy, α-hydroksykwasy lub mocznik)środki Keratynolityczne w połączeniu z kwasem salicylowymtopowe retinoidyw noworodkach i małych dzieciach należy stosować nośnik bez składników aktywnych. Unikać mocznika, kwasu salicylowego i kwasu mlekowego ze względu na ryzyko wchłaniania ogólnoustrojowego |
| leczenie doustne | doustne retinoidy (acitretyna lub izotretynoina) |
| inne środki | obserwacja ektropionu przez okulistę regularne oczyszczanie ucha zewnętrznego przez specjalistę ds.Unikanie uciążliwych czynności w wysokiej temperaturze otoczenia hydroterapia |
Kąpiel i mechaniczna eliminacja łusek
codzienna kąpiel jest zalecana dla pacjentów z ARCI w celu mechanicznego wyeliminowania łusek i śladów nawilżenia. Jest to łatwiejsze, jeśli pacjent jest zanurzony w wodzie na 15 do 30 minut. Niektórzy autorzy zalecają dodanie wodorowęglanu sodu do kąpieli, aby denaturalizować keratyny i sprawić, że woda będzie alkaliczna, a więc ułatwi eliminację łusek.129 inne produkty, które można dodać, to skrobia pszenna, skrobia kukurydziana lub skrobia ryżowa. Olejki do kąpieli nie są odpowiednie, ponieważ mogą prowadzić do niedrożności, a następnie ryzyka proliferacji bakterii i pogorszenia termoregulacji.
leczenie miejscowe
Nawilżacze i miejscowe środki keratolityczne są zwykle pierwszą opcją terapeutyczną. Poprawiają funkcje barierowe skóry i ułatwiają złuszczanie. Mogą wystąpić łagodne, miejscowe działania niepożądane, takie jak przemijający świąd, podrażnienie lub uczucie kłucia.
chlorek sodu, mocznik, octan witaminy E, glicerol i wazelina mogą być stosowane jako środki nawilżające i smarne. U pacjentów z grubą skalą i wyraźną hiperkeratozą można dodać 1 lub więcej leków keratolitycznych,takich jak α-hydroksykwasy (kwas mlekowy i glikolowy), 130 kwas salicylowy,N-acetylocysteina,131-133 mocznik (>5%), 134 i glikol propylenowy. Stosuje się również modulatory różnicowania keratynocytów. Należą do nich miejscowe retinoidy (tretinoina, adapalen,tazaroten), 135,136 kalcypotriol, 137 i dekspantenol.Miejscowe retinoidy często powodują podrażnienie i małe, bardzo bolesne pęknięcia.Ponadto istnieje ryzyko wchłaniania i działania teratogennego u płodnych kobiet, jeśli są one stosowane zbyt szeroko.138 w celu zwiększenia skuteczności środków keratolitycznych i nawilżających, opatrunek okluzyjny może być stosowany w określonych obszarach opornych na leczenie.Działanie addytywne lub synergiczne można również osiągnąć poprzez połączenie 2 lub więcej środków keratolitycznych lub nawilżających.140-142 leczenie powinno być zoptymalizowane dla każdej osoby, ze względu na wysoce zmienny charakter stanu i wrażliwości skóry oraz różnice w odpowiedzi na każdy zabieg. Proces optymalizacji może być wspomagany przez traktowanie jednej strony ciała inaczej niż drugiej, aby umożliwić porównania. Noworodki i małe dzieci powinny być leczone nośnikiem bez żadnych substancji czynnych, ponieważ skóra jest bardzo delikatna i wrażliwa, a większość środków keratolitycznych nie jest tolerowana. Ponadto ryzyko wchłaniania przez skórę produktów stosowanych miejscowo, takich jak mocznik, kwas salicylowy i kwas mlekowy, jest większe.143-145
leczenie ogólnoustrojowe
doustne retinoidy mają działanie keratolityczne, które pomagają wyeliminować skale i zapobiegają nadmiernej hiperkeratozie. Zarówno izotretynoina, jak i aromatyczne retinoidy (acitretyna i etretynian) okazały się skuteczne w leczeniu ARCIs.128,146,147 Acitretyna w dawce 0,5 do 1 mg / kg/d jest najczęściej stosowanym lekiem, szczególnie u pacjentów z LI. 148 pacjentów z CIE może mieć bardziej kompletną odpowiedź i przy niższych dawkach.
głównymi działaniami niepożądanymi są zaburzenia śluzowo-skórne, teratogenność, zaburzenia mięśniowo-szkieletowe oraz nieprawidłowy profil lipidów i zwiększenie aktywności aminotransferaz.149-152 w odniesieniu do działania teratogennego, w przypadku etretynianu i acitretyny należy unikać stosowania leków w czasie ciąży, a pacjentki powinny unikać zajścia w ciążę przez 3 lata po zakończeniu leczenia.Izotretynoina ma krótszy okres półtrwania i jest całkowicie eliminowana z organizmu po 1 miesiącu, więc może być preferowaną opcją u kobiet, które chcą zajść w ciążę.
monitorowanie leczenia powinno obejmować badanie laboratoryjne z testami czynnościowymi wątroby i profilem lipidowym przed rozpoczęciem leczenia, a następnie po 1 miesiącu i co 3 miesiące po rozpoczęciu leczenia. U kobiet w ciąży w okresie 2 tygodni przed rozpoczęciem leczenia należy wykonać test ciążowy, a od 4 tygodni przed rozpoczęciem leczenia do 3 lat po zakończeniu leczenia należy stosować skuteczną antykoncepcję (w przypadku acitretyny). Jeśli konieczne jest długotrwałe leczenie retinoidami, należy monitorować wzrost i rozwój kości. Niektórzy autorzy sugerują przeprowadzenie badania kości przed leczeniem, a następnie coroczne badanie.151 najnowsze wytyczne nie zalecają wykonywania rutynowej radiografii ze względu na możliwe szkodliwe skutki.Zamiast tego zaleca się selektywne badania radiologiczne u pacjentów z nietypowym bólem kości.
alternatywą dla ogólnoustrojowego leczenia retinoidami jest stosowanie leków znanych jako leki blokujące metabolizm kwasu retinowego, które zwiększają endogenne poziomy kwasu retinowego. Jednym z takich leków jest liarozol, który otrzymał status sierocy w leczeniu LI, CIE i HI przez Europejską Agencję Leków i Amerykańską Agencję Żywności i Leków.153-155 lek ten okazał się bardziej skuteczny niż acitretyna w badaniach klinicznych, a także jest lepiej tolerowany i ma lepszy profil farmakokinetyczny.154
Inna opieka medyczna
u pacjentów z ektropionem stosowanie sztucznych łez i smarów do oczu oraz nawilżanie skóry twarzy i policzków w szczególności może zmniejszyć cofanie powiek. Korekcja chirurgiczna jest ważną opcją w ciężkich przypadkach, ale zwykle musi być powtórzona kilka lat później. Hydroterapia może być korzystna.Należy zalecić pacjentom unikanie intensywnej aktywności fizycznej, gdy temperatura otoczenia jest wysoka, ponieważ hypohidrosis niesie ze sobą ryzyko udaru cieplnego i drgawek. Doustne retinoidy mogą poprawić termoregulację.157 fizjoterapia jest ważna w zapobieganiu przykurczom zgięciowym, szczególnie w przypadku HI. Regularne oczyszczanie zewnętrznego kanału słuchowego przez specjalistę od uszu, gardła i nosa może zapobiec gromadzeniu się skal, a tym samym zapobiegać utracie słuchu.
poradnictwo genetyczne i diagnostyka prenatalna
gdy u pacjenta zdiagnozowano rybią łuskę, należy mu zaoferować odpowiednie poradnictwo genetyczne, w którym wyjaśniono charakter zaburzenia, tryb transmisji i ryzyko przyszłych objawów w rodzinie. Diagnoza prenatalna może wskazywać, czy płód jest dotknięty, a jeśli tak jest, można zaoferować psychologiczne przygotowanie rodziny i przewidywane problemy podczas ciąży i porodu. Rodzice mogą mieć możliwość aborcji, jeśli leczenie nie jest dostępne. Ponadto, jeśli terapia genowa dla tych warunków stanie się dostępna w przyszłości, diagnoza prenatalna umożliwi zastosowanie tej terapii tak wcześnie, jak to możliwe.
przez ponad 20 lat diagnostykę prenatalną wykonywano poprzez pobranie próbki biopsji skóry płodu i zbadanie jej za pomocą mikroskopii optycznej, mikroskopii elektronowej lub immunohistochemii.158 159 ten inwazyjny zabieg można było wykonać tylko w późnych fazach ciąży, między 15 a 23 tygodniem ciąży i wiązał się z 1% do 3% ryzykiem utraty płodu.160,161 identyfikacja molekularnych mechanizmów dziedzicznych zaburzeń skóry umożliwiła znacznie wcześniejszą diagnozę opartą na technikach genetycznych.102,162-164 DNA płodu uzyskuje się przez amniopunkcję wykonaną między 15 a 20 tygodniem lub przez pobranie próbek kosmków kosmówkowych między 10 a 12 tygodniem. Ryzyko utraty płodu za pomocą tych technik jest mniejsze niż od 0,5% do 1%.Inne nieinwazyjne metody w rozwoju to analiza DNA komórek płodu i wolnego DNA płodu w krążeniu matek166, a także zastosowanie trójwymiarowych ultradźwięków.167,168
genetyczna diagnoza przedimplantacyjna może być również możliwa w technikach zapłodnienia in vitro, tak że tylko zapłodnione jaja wolne od mutacji są wszczepiane do macicy, co w większości przypadków pozwala uniknąć konieczności aborcji.169
przyszłe strategie genetycznego leczenia rybiej łuski
chociaż poczyniono istotne postępy w diagnostyce genetycznej rybiej łuski, nowe strategie są również realizowane w przypadku tych chorób.170 skóra jest najbardziej dostępnym organem do terapii transferu genów, więc takie techniki są minimalnie inwazyjne.171 jednak skóra ma również unikalne właściwości immunologiczne, które nie sprzyjają długotrwałej ekspresji produktu transgenicznego.172 In LI, w procesie transferu genów ex vivo udało się przywrócić prawidłową ekspresję TGM1 i skorygować fenotyp skóry przeszczepionej na plecach myszy z immunosupresją.173 174 Ostatnio odzyskano również fenotyp hodowanych keratynocytów u pacjentów z HI z powodu mutacji w genie ABCA12.3
konflikty interesów
autorzy oświadczają, że nie występują w nich konflikty interesów.