Frontiers in Neurology
wprowadzenie
Neuroakantocytoza jest nadrzędnym terminem dla grupy rzadkich zespołów, które charakteryzują się objawami neurologicznymi w połączeniu z kolczastymi zdeformowanymi krwinkami czerwonymi (akantocytami). Niniejszy raport koncentruje się na pląsawicy-akantocytozie (Chac) , która jest chorobą sierocą z szacowanymi 1000 dotkniętymi osobami na całym świecie spowodowanymi przez autosomalne recesywne mutacje w genie vps13a (wakuolar protein sorting 13 homolog A) na chromosomie 9Q (1). Choroba przebiega progresywnie, przyczyny zwiększonej śmiertelności mogą być zmniejszone funkcje motoryczne skorelowane z warunkami ryzyka, takimi jak dysfagia, ale opisano również nagłe nieoczekiwane zgony (2, 3). Do tej pory nie ma leczenia sprawczego.
podejrzane współistnienie akantocytozy i zaburzeń ruchowych zostało po raz pierwszy zgłoszone w latach 70.przez Levine ‘ a i Critchleya (4, 5) i od tego czasu zostało zaakceptowane jako główna cecha choroby. Jednak w naszym opisie przypadku opisujemy Rzadki fenotyp kliniczny ChAc bez oczywistych zaburzeń ruchowych, sugerując szerszą różnorodność prezentacji klinicznych. Narzędzia diagnostyczne muszą sprostać tym wyzwaniom, aby ustalić prawidłową diagnozę, proponujemy tutaj neuroobrazowanie molekularne jako kluczową metodę.
opis przypadku
po raz pierwszy u pacjenta w wieku 25 lat stwierdzono dwa nieprowokowane obustronne toniczne napady kloniczne. Nie było żadnej historii medycznej. Rezonans magnetyczny mózgu i EEG w tym wieku były w normie i nie rozpoczęto leczenia z powodu niechęci pacjenta i rzadkich zdarzeń. Jednak u pacjenta występowały napady padaczkowe, które obecnie występują jako padaczka częściowa. Opisał powtarzające się uczucie nagłych zawrotów głowy, które zostały zdiagnozowane jako zawrotna aura. Co więcej, miał napady dyscognitive, w których wykazywał albo (a) brak reakcji i recytowanie tureckich modlitw, (b) automatyzację ustną lub (c) Majsterkowanie rękami i wypowiadanie dźwięków—wszystkie częściowo ewoluowały w toniczne napady kloniczne. Wideo-EEG pokazało lokalne kompleksy spike-wave zarówno w prawym, jak i lewym płacie skroniowym. Zgodnie z semiologią napadów padaczkowych postawiono diagnozę obustronnej padaczki płata skroniowego i rozpoczęto leczenie lewetyracetamem.
w wieku 31 lat napady drgawkowe nie zostały całkowicie stłumione, w ramach dalszej diagnostyki padaczki płata skroniowego pacjent przeszedł FDG-PET (ryc. 1), który wykazał obustronny hipometabolizm meziotemporalny, zgodnie z pochodzeniem napadu meziotemporalnego. Stwierdzono jednak również wyraźny, bardzo nietypowy hipometabolizm prążkowia, który wzbudził podejrzenie neurodegeneracyjnego zaburzenia ruchu. W związku z tym rozpoczęto szeroko zakrojoną ocenę działań następczych (zob. wykres 2). Przeprowadzono badanie FP-CIT-SPECT, które wykazało umiarkowanie obustronną utratę dostępności transportera dopaminy w prążkowiu, zgodnie ze spadkiem integralności nigrostriatalnej (ryc. 1). W obrazie MRI o wysokiej rozdzielczości przedstawiono obustronny zanik jądra ogoniastego (ryc. 3). Badanie neurologiczne wykazało dyskretny, związany wzór chodu i hipomimię, ale nie ma innych objawów pozapiramidowego układu ruchowego i żadnych objawów opuszkowych, takich jak dystonia pokarmowa. Poza niskim stanem odruchów kończyn dolnych, u których badania przewodnictwa nerwowego potwierdziły sensorimotorową polineuropatię aksonalną, badanie neurologiczne było prawidłowe. Objawy neuropsychologiczne obejmowały aspekty zaburzenia lękowego, a pacjent zgłaszał trudności z utratą pamięci krótkotrwałej. Badania neuropsychologiczne nie potwierdziły jednak oczywistego nieprawidłowego działania mnestycznego, a raczej niespecyficznego rozproszenia, które mogło być dodatkowo wzmocnione przez niewystarczające tłumienie napadów w tym czasie. Wyniki badań laboratoryjnych były ujemne na obecność padaczki objawowej (tj., przeciwciała autoimmunologiczne-zapalenie mózgu, przeciwciała antyneuronalne). Płyn mózgowo-rdzeniowy nie wykazywał objawów zapalenia, ale umiarkowany wzrost białka (765 mg / l). Analiza amin biogennych w płynie mózgowo-rdzeniowym wykazała zwiększenie stężenia glutaminy, co wiązaliśmy z niedawnymi napadami. We krwi obwodowej stwierdzono 0,4% akantocytów. Testy serologiczne na chorobę Wilsona były negatywne. Zauważalne było utrzymujące się podwyższenie aktywności kinazy kreatynowej (zakres: 1000–3000 U / L), co doprowadziło do podejrzenia rozpoznania zaburzenia mitochondrialnego. W przypadku dalszych badań wykonano test mleczanu niedokrwiennego, który wykazał prawidłowe wyniki. Wykonano biopsję mięśnia M. gastrocnemicus, ale analiza czynności mitochondriów i łańcucha oddechowego była prawidłowa.
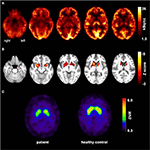
Rysunek 1. Wyniki badań obrazowania molekularnego. Przezosiowe obrazy FDG-PET (a) wykazały nie tylko łagodny obustronny hipometabolizm meziotemporalny (zgodnie z pochodzeniem napadu meziotemporalnego), ale także wyraźny hipometabolizm prążkowia, który okazał się bardzo znaczący w porównaniu ze zdrowymi kontrolami . Dodatkowe badanie FP-CIT-SPECT (C) wykazało również umiarkowaną utratę transporterów dopaminy w prążkowiu, świadcząc o spadku integralności nigrostriatalnej (dla porównania przedstawiono zdrową kontrolę dopasowaną do wieku; DVR, współczynnik objętości dystrybucji).

Rysunek 2. Przebieg kliniczny i diagnostyczny pacjenta indeksu.
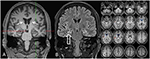
Rysunek 3. MRI pokazuje dyskretny obustronny zanik głowy Ogoniastej i Mniejszy i hiperintensywny prawostronny hipokamp wskazujący na prawostronne stwardnienie hipokampa . Lewy hipokamp jest nieco hiperintensywny, ale nie zanikowy. Obustronny zanik głowy Ogoniastej potwierdzono analizą CVR, w której niebieskie kolory wykazują zmniejszoną szarą i czerwoną barwę zwiększoną objętość płynu mózgowo-rdzeniowego, odpowiednio (C).
historia rodziny ujawniła pokrewieństwo jego rodziców (pierwszych kuzynów), którzy sami nie mieli znaczącej historii medycznej. Z sześciorga rodzeństwa, dwóch (w wieku 20-30 lat) cierpiało już na napady ogólne (patrz rysunek 4). Dokumentacja medyczna jego rodzeństwa nie była dla nas dostępna.
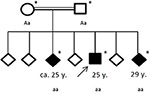
Rysunek 4. Rodowód rodziny dotkniętej chorobą, z wiekiem pierwszego napadu padaczkowego w latach (y). Arrow: patent index. Asterisk: przeprowadzono badania genetyczne. Aa, heterozygous; aa, homozygous dla c. 4326 T>A (P. Tyr1442*).
pod hipotezą dziedzicznego zaburzenia neurodegeneracyjnego ruchu, pacjent i jego rodzina zostali skierowani na badania genetyczne. Sekwencjonowanie Exome ujawniło homozygotyczną mutację novel truncating c. 4326 T> A (P. Tyr1442*) w genie ChAc VPS13A u trzech dotkniętych rodzeństwa. Rodzice spokrewnieni byli heterozygotyczni. W sekwencjonowaniu exome nie zidentyfikowano żadnych innych wariantów ani mutacji.
Sprawiał wrażenie lekkiego sztywności tylko pod pachami prawej kończyny górnej i łagodnego bradykinezji kończyn górnych. Podczas leczenia lewetyracetamem i lakozamidem u pacjenta nie stwierdzono napadów padaczkowych.
dyskusja
w niniejszym przypadku wykazujemy, że ChAc może manifestować się klinicznie w padaczce płata skroniowego bez znaczących objawów motorycznych z powodu nowej odpowiadającej mutacji w genie VPS13A.
przebieg choroby zazwyczaj charakteryzuje się postępującym zaburzeniem ruchowym (w tym pląsawicą, dystonią, parkinsonizmem), zmianami poznawczymi i psychiatrycznymi oraz objawami miopatycznymi z serologicznymi markerami akantocytozy i hiperkemii. Napady padaczkowe były wcześniej odnotowywane jako objaw ChAc (6), ale zaburzenia ruchu nadal są uważane za główny objaw. Pojawiają się dowody na to, że padaczka, a dokładniej padaczka pochodząca z płata skroniowego, może być niedocenianym fenotypem ChAc. Peluso et al. zgłoszono pacjenta podobnego do naszego, który nawet w wieku 46 lat nie wykazywał zaburzeń ruchowych, prezentując wyniki neuroobrazowania wskazujące na zaangażowanie zwojów podstawnych (7). Zgodnie z tym, Scheid et al. opisał trzech pacjentów cierpiących na padaczkę i stwardnienie płata skroniowego (na podstawie badań MRI) jako główny objaw i zdiagnozowano u nich ChAc (8). Ponadto Mente et al. niedawno dostarczono wyniki autopsji pacjenta z ChAc, który wykazywał nie tylko zanik zwojów podstawnych, ale także stwardnienie hipokampa (9). Zgodnie z naszym przypadkiem, obustronne zaangażowanie hipokampa wydaje się być powszechnym stwierdzeniem (10, 11). Dokładny związek między napadami a stwardnieniem jest jednak nadal kwestią kury lub jaja. Korelująca rola choreiny w rozwoju strukturalnym hipokampa nie jest jeszcze w pełni poznana. Co ciekawe, w mysim modelu Chac zaobserwowano zmiany strukturalne, ponieważ ekspresja białka hipokampa receptora GABA (A) gamma 2 i jego zakotwiczającego białka choreiny była zwiększona (12).
w rutynie klinicznej ten fenotyp nadal może być trudny w znalezieniu prawidłowej diagnozy. Pierwszą istotną wskazówką w kierunku choroby neurodegeneracyjnej u naszego pacjenta był wyraźny spadek metabolizmu glukozy w prążkowiu i umiarkowany spadek integralności nigrostriatalnej odpowiednio na FDG-PET i FP-CIT-SPECT. Niewielki zbiór funkcjonalnych wyników obrazowania, oparty na opisie serii przypadków, pokazuje, że zmiany w metabolizmie i dysfunkcji dopaminergicznej występują głównie w jądrze ogoniastym i skorupie (13). MRI mózgu ujawniło zanik jądra ogona w trakcie choroby, co zostało opisane jako typowe odkrycie w ChAc (14). W związku z tym różne cechy neurologiczne zwykle obejmują pląsawicę, dystonię pokarmową, dyskinezy orofaciolingwistyczne i Tiki, wraz z innymi objawami zwojów podstawnych, takimi jak parkinsonizm i dystonia (3). Kuszące jest spekulowanie, że obserwowane zmiany prążkowia u naszego pacjenta były nadal poniżej progu powodowania objawów (tj. wyników obrazowania prodromalnego), które ostatecznie mogą wystąpić w miarę postępu choroby. Dlatego zachęcamy do obrazowania strukturalnego i molekularnego jako wrażliwego narzędzia diagnostycznego w tej chorobie sierocej.
w ChAc istnieją opisy liczby akantocytów między 5 a 50% (3), ale istnieje kilka przypadków, które pokazują, że akantocyty mogą pojawić się dopiero późno w przebiegu choroby (15), mogą być bardzo niskie, a nawet całkowicie nieobecne (16). Dlatego akantocyty nie powinny być uważane za obowiązkowe do postawienia diagnozy. Dodatkowo padaczka może opóźnić diagnozę, ponieważ maskuje inne typowe objawy ChAc. W szczególności objawy psychiatryczne, takie jak zmiany osobowości i pogorszenie funkcji poznawczych, które są bardzo częste, mogą być błędnie interpretowane i przypisywane do działań niepożądanych związanych z lekiem (np. lewetyracetamu) lub związanych z napadami drgawkowymi. Wtórna hiperkemia jest obserwowana po ogólnych tonicznych napadach klonicznych, a uporczywe podwyższenie może być przeoczone.
nasz przypadek podkreśla również kluczową korzyść z sekwencjonowania exome w celu ustalenia prawidłowej diagnozy, zwłaszcza w rzadkich chorobach lub gdy objawy są niejasne. Rodzina genów ssaków vps13 (A-D) cieszy się coraz większym zainteresowaniem i zidentyfikowano mutacje w innych chorobach neurologicznych, takich jak autosomalna recesywna choroba Parkinsona lub autosomalna recesywna ataksja spinocerebellar (17). Recesywne mutacje w VPS13D powodują zaburzenia ruchowe w dzieciństwie (18). Podstawowa patofizjologia ChAc nie została jeszcze w pełni rozszyfrowana, ale wykazano, że VPS13A koduje białkową pląsawicę, która ulega ekspresji wszechobecnej w mózgu i w ogromnej liczbie innych tkanek (19). Badania ujawniają, że uczestniczy w szlakach sygnałowych, które regulują architekturę cytoszkieletu, egzocytozę i przetrwanie komórek (20). Inną mutacją opisaną jako występująca z fenotypem zdominowanym przez napady u 9 pacjentów z ChAc jest mutacja c. 2343del w genie VPS13A (21). Uzasadnione jest założenie, że specyficzne mutacje w tym samym Genie powodują pewną aberrację pląsawicy, która powoduje unikalny fenotyp, na przykład przez wpływanie na różne obszary mózgu. Jednak podstawowa patofizjologia jest nadal tylko spekulacyjna i dalsza dokumentacja mutacji sprawczych będzie interesująca. Pokazujemy tutaj, że nie opisana wcześniej mutacja okrojająca c.4326 T>A (P. Tyr1442*) powoduje podobny fenotyp z dominującą padaczką u wszystkich dotkniętych nią członków rodziny.
Uwagi końcowe
padaczka (szczególnie obustronna padaczka płata skroniowego) jest prawdopodobnie dominującym fenotypem ChAc. Dlatego należy starannie przeprowadzić wyszukiwanie dodatkowych czerwonych flag (takich jak hiperckemia, historia rodzinna, polineuropatia). W podejrzanych przypadkach należy szukać akantocytów we krwi i MRI mózgu, ponieważ narzędzia te są powszechnie dostępne. W razie wątpliwości diagnostykę należy uzupełnić FDG-PET i/lub FP-CIT-SPECT, ponieważ są one wrażliwe na wykrywanie zajęcia prążkowia. W autosomalnych padaczkach recesywnych ze wskazaniami na chorobę neurodegeneracyjną VPS13A należy następnie zastosować test pojedynczego genu lub pląsawicę Western blot w celu ustalenia właściwej diagnozy. Te ostatnie należy również uwzględnić w innych zaburzeniach neurodegeneracyjnych bez genetycznie zdefiniowanej etiologii.
Oświadczenie o etyce
pacjent uzyskał pisemną świadomą zgodę na udział w badaniu. Pacjent uzyskał pisemną, świadomą zgodę na opublikowanie niniejszego raportu przypadku.
JW, MR i SK brały udział w zarządzaniu pacjentami. JW napisał pierwszy szkic rękopisu. LF, HU i PM przygotowały dane liczbowe. Wszyscy autorzy przyczynili się do rewizji manuskryptu, przeczytali i zaakceptowali nadesłaną wersję.
Oświadczenie o konflikcie interesów
HU jest udziałowcem Veobrain Gmbh, spółki wydzielonej z Uniwersyteckiego Centrum Medycznego we Fryburgu.
pozostali autorzy oświadczają, że badania zostały przeprowadzone przy braku jakichkolwiek relacji handlowych lub finansowych, które mogłyby być interpretowane jako potencjalny konflikt interesów.
1. Walterfang M, Evans a, Leong Looi JC, Jung HH, Danek A, Walker RH, et al. Neuropsychiatria zespołów neuroakantocytozowych. Neurosci Biobehav Rev. (2011) 35: 1275-83. doi: 10.1016 / j.neubiorev.2011.01.001
PubMed Streszczenie / CrossRef Pełny Tekst / Google Scholar
2. Walker RH. Postępowanie w zespołach neuroakantocytozy. Drżenie Innych Hiperkinet. Mov. (2015) 5:346. doi: 10.7916 / D8W66K48
PubMed Abstract / CrossRef Full Text / Google Scholar
3. Velayos Baeza a, Dobson-Stone C, Rampoldi L, Bader B, Walker Rh, Danek A, et al. Pląsawica-Akantocytoza. Generaeviews®. Seattle, WA: University of Washington (1993).
4. Critchley EM, Clark DB, Wikler A. Akantocytoza i zaburzenia neurologiczne bez Betalipoproteinemii. Arch Neurol. (1968) 18:134–40.
PubMed Abstract
5. Levine IM, Estes JW, Looney JM. Dziedziczna choroba neurologiczna z akantocytozą. Nowy syndrom. Arch Neurol. (1968) 19:403–9.
PubMed Abstrakt / Google Scholar
6. Hardie RJ, Pullon HW, Harding AE, Owen JS, Pires M, Daniels GL, et al. Neuroakantocytoza. badanie kliniczne, hematologiczne i patologiczne 19 przypadków. Brain (1991) 114(Pt 1A):13-49.
PubMed Abstrakt / Google Scholar
7. Peluso S, Bilo L, Esposito M, Antenora A, Rosa ADR, Pappata s, et al. Pląsawica-akantocytoza bez pląsawicy: rozszerzenie fenotypu klinicznego. Choroba Parkinsona. (2017) 41:124–6. doi: 10.2016 / j.parkreldis.2017.05.013
PubMed Streszczenie / CrossRef Pełny Tekst / Google Scholar
8. Scheid R, Bader B, Ott DV, Merkenschlager a, Danek A. rozwój padaczki płata skroniowego w pląsawicy pląsawiczo-akantocytozie. Neurology (2009)73: 1419-22. doi: 10.1212 / WNL.0b013e3181bd80d4
PubMed Streszczenie / CrossRef Pełny tekst / Google Scholar
9. Mente K, Kim SA, Grunseich C, Hefti MM, Crary JF, Danek A, et al. Stwardnienie hipokampa i padaczka płata skroniowego w pląsawicy-akantocytozie: przypadek z oceną kliniczną, patologiczną i genetyczną. Neuropathol Appl Neurobiol. (2017) 43:542–6. doi: 10.1111 / nan.12403
PubMed Streszczenie / CrossRef Pełny Tekst / Google Scholar
10. Bader B, Vollmar C, Ackl N, Ebert a, la Fougère C, Noachtar S, et al. Obustronna padaczka płata skroniowego potwierdzona wewnątrzczaszkowym EEG w pląsawicy-akantocytozie. [2011-02-20 20: 34] doi: 10.1016 / j.2010.12.007
PubMed Streszczenie / CrossRef Pełny Tekst / Google Scholar
11. Marson AM, Bucciantini E, Gentile E, Geda C. Neuroacanthocytosis: clinical, radiological, and neurofizjological findings in an Italian family. Neurol Sci. (2003) 24:188–9. doi: 10.1007 / s10072-003-0123-1
PubMed Abstract / CrossRef Pełny tekst / Google Scholar
12. Kurano Y, Nakamura m, Ichiba m, Matsuda m, Mizuno e, Kato m, et al. Niedobór pląsawicy prowadzi do zwiększenia regulacji receptora GEFYRYNY i GABA(A). Biochem Biophys Res Commun. (2006) 351:438–42. doi: 10.1016 / j.bbrc.2006.10.070
PubMed Streszczenie / CrossRef Pełny Tekst / Google Scholar
13. Ehrlich DJ, Walker Rh. Funkcjonalne neuroobrazowanie i pląsawica: przegląd systematyczny. J Clin MOV Disord. (2017) 4:8. doi: 10.1186 / s40734-017-0056-0
PubMed Abstract / CrossRef Pełny tekst / Google Scholar
14. Connolly BS, Hazrati L-N, Lang AE. Wyniki neuropatologiczne w pląsawicy-akantocytozie: nowe spojrzenie na mechanizmy leżące u podstaw parkinsonizmu i drgawek. Acta Neuropathol. (2014) 127:613–5. doi: 10.1007 / s00401-013-1241-3
PubMed Abstract / CrossRef Pełny tekst / Google Scholar
15. Sorrentino G, De Renzo a, Minillo S, Nori o, Bonavita V. późne pojawienie się akantocytów w przebiegu pląsawicy-akantocytozy. J Neurol Sci. (1999) 163:175–8.
PubMed Abstrakt / Google Scholar
16. Bayreuther C, Borg M, Ferrero-Vacher C, Chaussenot A, Lebrun C. Choréo-akantocytoza bez akantocytów. Revue Neurol. (2010) 166:100–3. doi: 10.1016 / j.neurol.2009.03.005
CrossRef Pełny Tekst / Google Scholar
17. Lesage S, Drouet V, Majounie E, Deramecourt V, Jacoupy m, Nicolas A, et al. Utrata funkcji VPS13C w Autosomalno-recesywnym Parkinsonizmie powoduje dysfunkcję mitochondriów i zwiększa mitofagię zależną od PINK1/Parkina. Am J Hum Genet. (2016) 98:500–13. doi: 10.1016 / j.ajhg.2016.01.014
PubMed Streszczenie / CrossRef Pełny Tekst / Google Scholar
18. Gauthier J, Meijer IA, Lessel D, Mencacci NE, Krainc D, Hempel m, et al. Recesywne mutacje w >VPS13D powodują zaburzenia ruchowe w dzieciństwie. Ann Neurol. (2018) 83:I6. doi: 10.1002 / ana.25204
PubMed Streszczenie / CrossRef Pełny Tekst / Google Scholar
19. Kurano Y, Nakamura m, Ichiba m, Matsuda m, Mizuno e, Kato m, et al. Dystrybucja in vivo i lokalizacja pląsawicy. Biochem Biophys Res Commun. (2007) 353:431–5. doi: 10.1016 / j.bbrc.2006.12.059
PubMed Streszczenie / CrossRef Pełny Tekst / Google Scholar
20. Lang F, Pelzl L, Schöls L, Hermann a, Föller M, Schäffer TE, et al. Neurony, erytrocyty i nie tylko-różnorodne funkcje pląsawicy. 25:117-26. doi: 10.1159/000485457
PubMed Abstract / CrossRef Pełny tekst / Google Scholar
21. Benninger F, Afawi Z, Korczyn AD, Oliver KL, Pendziwiat m, Nakamura M, et al. Napady padaczkowe jako prezentujący i widoczny objaw w pląsawicy-akantocytozie z mutacją genu c. 2343DEL VPS13A. (2016) 57:549-56. / align = “left” /10111113318
PubMed Abstract | CrossRef Full Text | Google Scholar