Frontiers in Pediatrics
- wprowadzenie
- zespół długiego QT
- Lqts1
- LQTS2
- LQTS3
- LQTS4
- Lqts5
- lqts6
- LQTS7
- LQTS8
- Lqts9 i Lqts10
- LQTS11
- Lqts12
- LQTS13
- nabyte LQTS
- cechy elektrokardiograficzne w trzech typowych postaciach zespołu długiego QT
- genotyp-fenotyp
- leczenie kliniczne LQTS
- LQTS: Perspektywa Saudyjska
- Oświadczenie o konflikcie interesów
- podziękowania
wprowadzenie
rodzinne lub dziedziczne zaburzenia rytmu serca obejmują znaczny odsetek zaburzeń rytmu serca, a także przyczynę nagłej śmierci sercowej (SCD) (1, 2). W ciągu ostatnich dwóch dekad naukowcy i klinicyści włożyli ogromny wysiłek w rozwikłanie skomplikowanych i złożonych mechanizmów wrodzonych rodzinnych zaburzeń rytmu serca (1-8). Aby zrozumieć mechanizm arytmogenezy, musimy znać podstawy struktury komórkowej serca i ich właściwości elektrofizjologiczne. Miocyty serca są głównymi funkcjonującymi komórkami w sercu i są one szeroko sprzężone, dzięki czemu impulsy propagują się szybko i równomiernie. Kardiomiocyty są oddzielone od siebie wyspecjalizowaną granicą zwaną dyskiem interkalowanym; białka złącza szczeliny, desmosomy serca i kanały jonowe znajdują się w Dysku interkalowanym (9). Złącza szczelinowe składają się z ciasno upakowanych koneksyn, które umożliwiają międzykomórkową wymianę małych cząsteczek, a także umożliwiają przepływ prądów wzbudzających z jednej komórki do sąsiedniej komórki. Desmosomy wraz z przyłączami przylegającymi odpowiadają za mechaniczne przyłączenie poszczególnych kardiomiocytów. Wszystkie te składniki w dyskach interkalowanych są kolejno segregowane, a każdy składnik pełni swoją unikalną funkcję; zakłócenie jednego składnika wpływa na funkcję innych składników, co predysponuje serce do rozwoju arytmii (9-11). Kanały jonowe serca są kompleksami białkowymi tworzącymi pory, które zapewniają napięciowy i misternie skoordynowany ruch do wewnątrz i na zewnątrz prądów jonowych przez błony komórkowe, niezbędny do generowania i propagacji rytmu serca. Zespół długiego QT (lqts), zespół krótkiego QT (SQTS), zespół chorego Zatoki (SSS), wada przewodzenia serca (CCD), BrS, polimorficzny częstoskurcz komorowy katecholaminergiczny (CPVT), wczesny zespół repolaryzacji (ERS) i rodzinne migotanie przedsionków (AF) są obecnie znanymi kanałopatiami serca, które mogą wystąpić z powodu pojedynczej lub wielokrotnej wady genów związanych z generowaniem i propagacją rytmu serca.
w tym przeglądzie opiszemy wyłącznie arytmie związane z LQTS, jej patofizjologię i obecnie dostępne leczenie kliniczne. Jesteśmy pionierami w wyjaśnianiu patologii genetycznej w rodzinnych arytmiach serca w Arabii Saudyjskiej (1, 12-14), nadal prowadzimy badania kardiogenetyczne w Arabii Saudyjskiej, pod koniec tego przeglądu omówimy obecnie dostępną wiedzę na temat genetycznych i klinicznych wyników uzyskanych od kilku rodzin saudyjskich z historią omdleń i SCD. Dodamy również niedawno opublikowane dane na temat LQT od zespołu z King Faisal Specialist Hospital and Research Centre, Rijad (15).
zespół długiego QT
wrodzone LQTS jest zaburzeniem dziedzicznym, definiowanym przez wydłużenie odstępu QT na elektrokardiogramie (EKG). Pacjenci ze wszystkimi postaciami LQT są predysponowani do tachyarytmii komorowej, torsades de pointes (TDP) prowadzącej do nawracających omdleń lub SCD. W wielu przypadkach omdlenia lub nagła śmierć mogą być pierwszą i jedyną manifestacją. LQTS dotyka około 1 Na 2000 osób na całym świecie (16). Cechą charakterystyczną LQTS jest wydłużenie odstępu QT w EKG (skorygowane o częstość akcji serca, tj. QTc). Prawidłowe wartości QTc wynoszą 440 ms u mężczyzn i 450 ms u kobiet. U dzieci istotne są wartości zależne od wieku i płci. Ostatnie zalecenia konsensusu dotyczące diagnozy LQTS są następujące (17, 18):
1. Lqts jest diagnozowany:
a. w obecności wskaźnika ryzyka lqts ≥3,5 przy braku wtórnej przyczyny wydłużenia odstępu QT i/lub
B. w obecności jednoznacznie patogennej mutacji w jednym z genów LQTS lub
c. W obecności odstępu QT skorygowanego o częstość akcji serca według wzoru Bazetta (QTc) ≥500 ms W powtarzanym 12-ołowiowym elektrokardiogramie i przy braku wtórnej przyczyny wydłużenia odstępu QT.
2. LQTS można zdiagnozować w obecności QTc między 480 a 499 ms w powtarzanych 12-odprowadzeniowych EKG u pacjenta z niewyjaśnionym omdleniem, przy braku wtórnej przyczyny wydłużenia odstępu QT i przy braku mutacji patogennej.
u pacjentów z czasem trwania odstępu QTc ≥500 ms skumulowane prawdopodobieństwo wystąpienia pierwszego omdlenia było istotnie większe niż u pacjentów z czasem trwania odstępu QTc <500 ms od urodzenia do 20 roku życia (19). Jednak u pacjentów, u których wystąpiły 2, 3 i 4 omdlenia, ryzyko wystąpienia kolejnego omdlenia było praktycznie identyczne u pacjentów z wąskim lub wydłużonym czasem trwania odstępu QTc (19). Molekularne podstawy LQTS są niejednorodne i do tej pory opisano mutacje w 13 różnych genach powodujące powstawanie LQTS (1-8, 19, 20). Wada genetyczna występuje zwykle u 70% pacjentów z LQTS w jednym z tych 13 genów (1-8, 19, 20). Wśród obecnie znanych 13 różnych typów LQTS, najczęściej są LQTS1, LQTS2 i LQTS3, z powodu wad genów kanału jonowego serca, odpowiednio KCNQ1, KCNH2 i SCN5A. Dziewięćdziesiąt procent wszystkich mutacji przyczynowych LQTS znajduje się w tych trzech genach (1-8, 19, 20). Mutacje w pozostałych 10 genach są rzadkie i stanowią tylko ∼10% wszystkich obecnie znanych mutacji LQTS.
zespół długiego QT jest zwykle chorobą autosomalną dominującą, ale czasami wielokrotne mutacje w jednym genie lub w różnych genach można znaleźć u 5-10% pacjentów z LQTS (21, 22). Pacjenci z wieloma mutacjami mogą wykazywać dłuższy odstęp QTc w porównaniu z pacjentami z pojedynczą mutacją i tacy pacjenci są również 3,5-krotnie bardziej narażeni na zagrażające życiu zdarzenia sercowe (21, 22)
Lqts1
mutacje w genie KCNQ1 są najczęstszą formą wszystkich LQTS i określane jako lqts typu 1 (LQTS1). Pięćdziesiąt procent wszystkich mutacji przyczynowych LQTS znajduje się w genie KCNQ1 (20). KvLQT1 (zwany także Kv7.1) jest białkiem wytwarzanym przez gen KCNQ1, a białko tworzy tetramery w retikulum endoplazmatycznym wewnątrz komórek, które putately współwystępuje z białkiem norki (kodowanym przez KCNE1) i są następnie transportowane do błony plazmatycznej miocytów serca, gdzie pośredniczą w powoli aktywującym prądzie, który przyspiesza repolaryzację potencjału czynnościowego w tkankach serca, prąd ten jest znany jako IKs (Fig. 1). Przyczynowe mutacje lqts1 kcnq1 są głównie mutacjami missense i w rzadkich przypadkach mogą być mutacjami z przesunięciem ramki w regionie C-terminalu (23). Zaburzenia rytmu serca u nosicieli mutacji KCNQ1 są wywoływane przez bodźce adrenergiczne, np. stres emocjonalny, wysiłek fizyczny, nurkowanie, pływanie (24-26). Pacjenci z mutacjami w domenach Przezbłonowych KvLQT1 są narażeni na większe ryzyko zdarzeń sercowych związanych z LQTS i mają większą wrażliwość na stymulację współczulną(26, 27). Pacjenci z mutacjami missense są narażeni na zwiększone ryzyko w porównaniu do pacjentów z mutacjami bezsensownymi lub obcinającymi (27, 28). Zmienność 3 ‘ – UTR w genie KCNQ1 wpływa również znacząco na wrażliwość na arytmię, przypuszczalnie poprzez wpływ na ekspresję genu (29). Pacjenci z arytmią spowodowaną mutacjami KCNQ1 reagują dość dobrze na β-blokery, ale niektórzy pacjenci nadal mogą być mniej wrażliwi lub nawet odporni na ten lek. W ostatnim artykule, Barsheshet et al. (30) twierdził, że pacjenci z mutacjami poza regionem pętli cytoplazmatycznej (C-loop) w kvlqt1 są mniej wrażliwi na β-blokery.
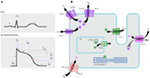
Rysunek 1. Prądy jonowe wpływające na potencjał czynnościowy komory (a) i schematyczne przedstawienie kardiomiocytów wykazujących (tylko) białka biorące udział w patogenezie dziedzicznych zespołów arytmii (B). W lit.a) potencjał działania jest dostosowany do jego przybliżonego czasu działania podczas EKG. W (B) nie przedstawiono ankyrin-B, białka adaptera biorącego udział w zespole długiego QT typu 4.
homozygotyczne lub złożone heterozygotyczne mutacje w genie KCNQ1, które mogą powodować recesywną postać choroby, zespół Jervella i Lange-Nielsena (Jlns), typ 1 (31), są dość rzadkie . Pacjenci z JLNS cierpią również na ciężkie zaburzenia rytmu serca i głuchotę (31, 32). Pacjenci z mutacjami kcnq1 powodującymi JLNS zwykle nie mają żadnych funkcjonalnych IKs (28, 31-33). Możliwe jest, że niektórzy pacjenci nie mogą mieć głuchoty pomimo homozygotycznych lub złożonych heterozygotycznych mutacji w KCNQ1, takie przypadki są określane jako autosomalne recesywne LQTS1 (13, 32). U tych pacjentów może nadal występować niewielka ilość czynnościowego prądu IKs (<10% całkowitego IKs), co utrzymuje funkcję słuchu, ale ich zaburzenia rytmu serca są równie poważne, jak u pacjentów z JLNS (13, 32).
LQTS2
ten typ LQTS jest równie rozpowszechniony jak LQTS1, co stanowi 35-40% pacjentów z lqts z wykrywalną mutacją (1, 20, 24, 25). KCNH2 koduje białko HERG (Kv11.1), które jest podjednostką α szybko aktywującego się opóźnionego prostownika prądu K+ (IKr). Mutacje patogenne w tym genie, które zmniejszają funkcję kanału Kv11.1, wydłużają czas trwania odstępu QT (rycina 1) i są przyczynowe dla LQTS2. Dwadzieścia dziewięć procent ataków omdlenia w LQTS2 występuje podczas odpoczynku/snu, a tylko 13% ataków omdlenia odnotowano podczas ćwiczeń (25, 34). Nagłe wstrząsające dźwięki, np. dzwonki budzika, dzwonki do drzwi, dzwonki telefonu, zazwyczaj wywołują ataki omdleń u tych pacjentów (24, 34). Pacjenci z mutacjami w regionie tworzenia porów kv11.1 (kodowanym przez gen KCNH2) są podatni na wysokie ryzyko zdarzeń sercowych związanych z arytmią w porównaniu z pacjentami z mutacjami w regionie bez porów (35). U noworodków blok przedsionkowo-komorowy 2: 1 jest preferencyjnie związany z mutacjami KCNH2 (36). Całkowity blok AV powikłany przez LQTS stwierdzono również U 17% dorosłych pacjentów z mutacją w genie KCNH2 (37). Homozygotyczne mutacje w KCNH2 są rzadkie i gdy występują, pacjenci cierpią na ciężką postać LQTS, z 2:1 blok AV i ciężkie komorowe zaburzenia rytmu serca, zarówno w stadiach wewnątrzmacicznych, jak i po urodzeniu (11, 38-40). Oprócz licznych mutacji opisanych w patologii LQTS2, wspólne warianty polimorficzne w genie KCNH2 mogą również modulować nasilenie choroby. Intrygującym przykładem jest polimorfizm K897T (SNP) w KCNH2, który występuje u 33% populacji ogólnej (41). Stwierdzono, że K897T nasila patogenność mutacji KCNH2 (42, 43).
LQTS3
Nav1.5 jest tworzącą pory podjednostką α zależnego od napięcia kanału sercowego na+, jest integralnym białkiem błonowym kodowanym przez gen SCN5A i bierze udział w inicjacji i przewodzeniu potencjałów akcji serca. Kanały na + serca składają się z tworzącej pory podjednostki α (kodowanej przez SCN5A) i jednej lub więcej pomocniczych podjednostek β.
LQTS3 jest spowodowany wzmocnieniem mutacji funkcyjnych, które zakłócają szybką inaktywację podjednostki α (Fig. 1), A mutację w genie SCN5A opisano u < 10% wszystkich pacjentów z LQTS z mutacją (1 ,20, 24). U pacjentów z LQTS3 większość (39%) zdarzeń sercowych wystąpiło podczas snu/odpoczynku (25), a około 13% zdarzeń wystąpiło podczas wysiłku fizycznego (25). W kilku przypadkach wykazano, że pojedyncza mutacja SCN5A wywiera dwa lub nawet trzy różne fenotypy arytmii w tej samej rodzinie, np. LQTS, BrS lub CCD (44-47). U pacjentów płci męskiej z mutacją LQTS3 objawy mogą rozwinąć się znacznie wcześniej niż u pacjentów płci żeńskiej(48). Zarówno heterozygotyczne, jak i homozygotyczne mutacje SCN5A zostały opisane w LQTS3 z funkcjonalnym blokiem AV 2:1 (49). Niedawno znaleźliśmy irańską rodzinę z mutacją 1507_1509delqkp u wielu członków rodziny, w której pacjenci mieli połączone LQTS i CCD (dane nie pokazane). Mutacja ta została zgłoszona u pacjentów z innych krajów, jak również, co sugeruje, że jest to powtarzające się i hot spot mutacji . Mutacje o takiej charakterystyce utraty i wzmocnienia funkcji w różnych fazach potencjału czynnościowego odnotowano również w mutacjach 1493delk i 1795insd (44, 51).
czasami SNP może również wywierać patogenny wpływ na swoich nosicieli. S1103Y jest powszechnym wariantem genu SCN5A, obecnym u 13% Afroamerykanów (52). Nosiciele z tym wariantem są narażeni na zwiększone ryzyko arytmii i zespołu nagłej śmierci niemowląt (SIDS) (52).
LQTS4
lqts4 reprezentuje pierwszą Nie-kanałową formę LQTS. Mutacja w ANK2, białku adaptera, prowadzi do wewnątrzkomórkowego przeciążenia wapnia, które przyczynia się do LQTS4 (53, 54). Oprócz wydłużenia odstępu QT u pacjentów z tym zespołem może występować bradykardia zatokowa, napadowy AF i CPVT (54). Patogenne działanie mutacji ANK2 może być umiarkowane do ciężkiego, a ekspresja kliniczna zależy od ciężkości mutacji.
Lqts5
mutacje w KCNE1 są związane z Lqts5 (Fig. Heterozygotyczne mutacje KCNE1 zmniejszają IKs, wywierając dominujący negatywny wpływ na towarzyszący mu normalny allel i prowadzą do opóźnionej repolaryzacji serca (ryc. 1), odpowiedzialnej za zwiększone ryzyko arytmii (6). Pacjenci z homozygotycznymi mutacjami KCNE1 cierpią na JLNS (typ 2) (57, 58).
D85N jest polimorfizmem w genie KCNE1, obecnym w 0.7-1% populacji ogólnej (41). W badaniu Nishio et al. (59), polimorfizm D85N był częściej stwierdzany u pacjentów z LQTS, co czyni go genotypem ryzyka w patologii LQTS (potencjalnie tylko w populacji azjatyckiej). W Europie D85N zgłaszano u 5% pacjentów z nabytym LQTS (aLQTS) (w kohorcie 32 pacjentów), u których wystąpiła TdP (60).
lqts6
Gen KCNE2 koduje peptyd 1 związany z Norkami (MiRP1), przypuszczalną podjednostkę β kanału potasowego IKr serca (Fig. Mutacje w genie KCNE2 mogą również prowadzić do wad szybko aktywującego się składnika opóźnionego prostownika prądu potasowego (IKr), patologicznej podstawy LQTS6 (61). Bodźce słuchowe / akustyczne, takie jak hałas budzika, Dzwonek Do drzwi itp. może wywoływać ataki synkopalne u nosicieli mutacji kcne2, podobne do mutacji kcnh2 (62).
LQTS7
zespół ten jest również znany jako zespół Andersena–Tawila (ATS). ATS jest rzadkim zaburzeniem, objawiającym się sporadycznymi omdleniami i zatrzymaniem krążenia. Funkcje EKG obejmują łagodne wydłużenie odstępu QT, nieprawidłowe fale U, częstą ektopię komorową, dwukierunkowy częstoskurcz komorowy (VT) i polimorficzną VT. Zespół ten wykazuje również cechy pozasłoneczne, np. okresowe porażenie mięśni szkieletowych i problemy rozwojowe, takie jak rozszczep podniebienia, nisko osadzone uszy, niski wzrost i wady rozwojowe kończyn (63). U większości pacjentów z klinicznie rozpoznanym ATS stwierdzono mutację w KCNJ2 (63). KCNJ2 koduje podjednostkę tworzącą pory kanałów potasowych prostujących do wewnątrz (IK1) (Fig.1) (64, 65).
LQTS8
pacjenci, znani również jako zespół Timothy ‘ ego (TS), wykazują znaczne wydłużenie odstępu QT w EKG, które jest połączone z syndaktylią, łysieniem przy urodzeniu i małymi zębami w 100% przypadków oraz mniej penetrującymi wadami strukturalnymi serca, upośledzeniem umysłowym, autyzmem i cechami dysmorficznymi twarzy (66). Istnieją dwa podtypy: TS1 (klasyczna) i TS2 (rzadka forma).
TS2 jest kardiologicznie silniejszy niż TS1 (66, 67). Pacjenci z TS2 również nie mają syndaktylii (67). Mutacje w podjednostce α-1 prądu wapniowego typu L (ICA-l) kodującego Gen CACNA1C prowadzą do obu postaci TS (LQTS8). W genie CACNA1C występują alternatywnie splicowane, wzajemnie wykluczające się eksony-8, dla jasności są one nazywane eksonami – 8 i eksonem-8a. w sercu i mózgu, gdzie CACNA1C jest głównie wyrażany, ekson-8 znajduje się w ∼80% transkryptów mRNA, a ekson-8A jest obecny ∼20% transkryptów (66). Mutacja G406R w eksonie – 8A jest przyczynowa do klasycznej postaci TS (TS1), a g402s w eksonie-8 odnotowano w severerze w TS2. Nowym dodatkiem do tej listy jest mutacja Ala1473Gly, która została opisana u niemowlęcia TS z Dalej rozszerzonym fenotypem (68). Wszystkie mutacje były albo de novo, albo mozaikowe u rodzica, a cały wynik zyskał funkcję kanału ICa-L (66-69).
Lqts9 i Lqts10
mutacje w CAV3 lub SCN4B powodują wzmocnienie funkcji w późnym INa, powodując fenotyp podobny do lqts3 (70-74). Są one znane jako LQTS9 (związane z mutacją CAV3) i LQTS10 (związane z mutacją SCN4B).
Caveolae są ładnie opisane przez Engelmana i wsp. (72) jako “małe jaskinie” w błonie plazmowej. Są to małe, niepowlekane pestki i są uważane za miejsce ważnych dynamicznych i regulacyjnych zdarzeń w błonie plazmatycznej (72, 73). Caveoliny są głównymi białkami w caveolae, a caveolin-3 (kodowany przez gen CAV3) znajduje się w kardiomiocytach i komórkach mięśni szkieletowych. Kilka sercowych kanałów jonowych zostało specjalnie zgłoszonych do zlokalizowanych w caveolae wyekstrahowanych z miocytów serca, które są wzbogacone w caveolin-3 (72, 73). Dodatkowo składniki kaskady sygnałowej receptora β-adrenergicznego są również obecne w błonach wzbogaconych caveolae (72, 73).
SCN4B koduje NaVß4, który jest pomocniczą podjednostką β kanału sodowego serca. Do tej pory odnotowano tylko jedną mutację (L179F) w tym genie w meksykańskiej rodzinie z wieloma dotkniętymi członkami rodziny (74). Stwierdzono, że mutacja w tym genie powoduje zwiększenie funkcji prądu Nav1.5 (74).
LQTS11
w sercu współczulna regulacja czasu trwania potencjału czynnościowego serca (APD) odbywa się za pośrednictwem aktywacji receptora β-adrenergicznego (β-AR), co wymaga montażu AKAP9 (Yotiao) z podjednostką α (KvLQT1) kanału IKs. Mutacja w AKAP9 powoduje LQTS11 (75). Do tej pory odnotowano tylko jedną mutację, S1570L, w AKAP9 (75).
Lqts12
mutacja w genie α-1-Syntrofiny (SNTN1) jest przyczynowa dla LQTS12. Mutacja w tym genie prowadzi do wzmocnienia funkcji kanału sodowego serca (Nav1. 5), który jest patologiczną podstawą LQTS12 (76).
LQTS13
g sprzężona z białkiem, wewnętrznie prostująca podjednostka kanału potasowego (Kir3.4) jest kodowana przez gen KCNJ5. Mutacja utraty funkcji w tym genie może spowodować LQTS13 (77). Do tej pory tylko jedna mutacja, G387R, została opisana w Chińskiej rodzinie z dziewięcioma pacjentami z tą mutacją. Jako patologię LQTS u pacjentów sugerowano zmniejszenie ekspresji błony komórkowej Kir3.4.
nabyte LQTS
oprócz wrodzonych LQTS istnieje również inny wariant LQTS znany jako aLQTS, który jest spowodowany czynnikami i substancjami zmniejszającymi strumień potasu i upośledzającymi zdolność mięśnia sercowego do repolaryzacji. Dobrze rozpoznanymi warunkami są Płeć żeńska, hipokaliemia i leki hamujące kanały potasowe serca (78-80). Wiele powszechnie przepisywanych leków może również preferencyjnie wiązać i blokować kanał HERG (kv11.1, białko kodowane przez gen KCNH2) i predysponuje do alqt (79, 80). Niedawno wykazano, że blokada IKs może również przyczyniać się do alqt indukowanych lekami, szczególnie w przypadku naruszenia rezerwy repolaryzacji (81). Stwierdzono, że fluoksetyna i norfluoksetyna hamują właściwości IKs, zarówno in vivo, jak i in vitro i prowadzą do znaczącego LQTS (81). Polimorfizmy, D85N w norkach (Gen KCNE1), T8A, Q9E w MiRP1 (Gen KCNE2), które są przypuszczalnymi β-sub-jednostkami kanałów IKs i IKr, powodowały aLQTS (82, 83). Autoimmunologiczne LQTS zgłaszano również u pacjentów z IgG zawierającymi przeciwciała anty-HERG (84).
cechy elektrokardiograficzne w trzech typowych postaciach zespołu długiego QT
typowe wzorce fali ST-T są obecne u większości pacjentów z genotypowym LQTS i mogą być wykorzystane do identyfikacji genotypów lqts1, LQTS2 i prawdopodobnie lqts3 (85, 86).
postać LQTS1 lqts jest związana z szeroką falą T bez skrócenia odstępu QT podczas ćwiczeń (ryc. 2A). LQTS2 jest związany z falami t o niskiej amplitudzie, często dwufidowymi (rys. 2b). LQTS3 jest związany z długim segmentem izoelektrycznym i wąską, wysoką falą T (rys. 2C). Pauza zależność początku TDP w wrodzonych LQTS jest specyficzna dla genotypu, jest dominująca w LQTS2, ale prawie nieobecna w LQTS1 (87).

Rysunek 2. Zapis elektrokardiogramu od pacjentów z LQTS1, LQTS2 i LQTS3. A) 12-ołowiowe EKG 18-letniego mężczyzny z mutacją KCNQ1. Odstęp QT jest wydłużony (QTc = ±500 ms). Segment ST ma szeroką podstawę i stosunkowo dużą amplitudę. Interwał przewodzenia jest Normalny (standardowa kalibracja). B) 12-ołowiowe EKG 14-letniej dziewczynki z mutacją KCNH2. Odstęp QT jest wydłużony (QTc ± 520 ms). Segment ST jest karbowany w przewodzie v3 i ma stosunkowo niską amplitudę w przewodzie skrajnym. Interwał przewodzenia jest Normalny (standardowa kalibracja). C) 12-ołowiowe EKG 12-letniego chłopca z mutacją SCN5A. Odstęp QT jest wydłużony (QTc ± 600 ms). Segment ST ma długi (prawie) izoelektryczny segment z dużą, ostrą i wąską falą T. Interwał przewodzenia jest Normalny (standardowa kalibracja).
chociaż wzorce mogą sugerować specyficzny genotyp LQT, często spotykano wyjątki.
genotyp-fenotyp
zespół długiego QT jest chorobą autosomalnie dominującą. Analiza genotypu i fenotypu wśród heterozygotycznych nosicieli mutacji została przeprowadzona dość szeroko u pacjentów z LQTS1, lqts2 i lqts3. W okresie dzieciństwa ryzyko wystąpienia zdarzeń sercowych jest istotnie większe u mężczyzn z LQTS1 niż u kobiet z lqts1, podczas gdy u pacjentów z LQTS2 i LQTS3 (88-91) nie obserwowano istotnych różnic w ryzyku wystąpienia zdarzeń sercowych związanych z płcią. W wieku dorosłym (również po 40. roku życia), LQTS1 i lqts2 kobiety mogą mieć znacznie większe ryzyko zdarzeń sercowych niż odpowiedni mężczyźni (88-91). Ogólnie wydaje się, że letalność zdarzeń sercowych jest dominująca u pacjentów z LQTS3 niż u pacjentów z lqts1 i lqts2 (91). Kobiety z LQTS mają zmniejszone ryzyko wystąpienia zdarzeń sercowych w czasie ciąży, ale ryzyko to znacznie wzrasta w okresie 9 miesięcy po porodzie, szczególnie u kobiet z mutacją w genie KCNH2 (92).
nagła śmierć sercowa u dzieci może być również spowodowana mutacjami w genach kanału jonowego serca (93-96). Około 28% dzieci z niewyjaśnionym SCD (między 1 A 18 rokiem życia, średnia wieku: 12,3 ± 3,8 roku) było nosicielami mutacji w genach przyczynowych LQTS (97). W SIDS, mutacje w SCN5A wydawały się dominujące (98, 99), ale, mutacje w KCNQ1, KCNH2, KCNE2 i CAV3, SCN4B i SCN3B również stwierdzono (100, 101). Wewnątrzmaciczne zgony płodu odnotowano również z powodu wad w genach kanału jonowego serca (12, 102).
leczenie kliniczne LQTS
zaprzestanie stosowania wszystkich leków, o których wiadomo, że wydłużają odstęp QT, a także wyrównywanie zaburzeń równowagi elektrolitowej i (lub) przyspieszanie warunków metabolicznych, powinno być głównym celem leczenia pacjentów (nabytych) LQTS. Objawy w LQTS mają często podłoże adrenergiczne, dlatego ogólnie zaleca się ograniczenie udziału pacjentów w zajęciach sportowych (24, 25). Podstawą klinicznej terapii LQTS jest blokada β. Zwykle stosuje się długo działające preparaty propranololu, nadololu i metoprololu, a ich skuteczność w blokadzie β ocenia się poprzez stępienie tętna wysiłkowego (np. przez >20%) (24, 25). Wśród wszystkich β-blokerów propranolol i Nadolol są uważane za lepsze niż metoprolol u pacjentów z objawami (103). Ponadto blokada β może być również stosowana jako leczenie profilaktyczne u nosicieli cichych mutacji w celu zmniejszenia SCD (24). W badaniu przeprowadzonym przez Barsheshet et al. (30), pacjenci z mutacjami typu C-loop missense w genie KCNQ1 wykazywali wysokie ryzyko zagrażających życiu zdarzeń sercowych i odnosili znaczące korzyści z leczenia β-blokerami. Ze względu na to, że u kobiet z LQTS2 występuje zwiększone ryzyko w okresie 9 miesięcy po porodzie, należy przepisać β-adrenolityki w celu zmniejszenia zdarzeń sercowych w tym okresie wysokiego ryzyka (92). Wszczepialne kardiowertery-defibrylatory (ICDs) można rozważyć u pacjentów z nawracającymi omdleniami pomimo leczenia β-blokerem lub u pacjentów z wysokim ryzykiem zatrzymania krążenia (np., objawowe LQTS2 i lqts3 z udokumentowanym wydłużeniem odstępu QTc). LCSD) jest zalecane u pacjentów z LQTS wysokiego ryzyka, u których ICD jest przeciwwskazane lub odrzucone, a leki blokujące receptory β-adrenergiczne nie są skuteczne, nie są tolerowane, akceptowane lub przeciwwskazane (18, 104). Pacjenci z JLNS zwykle mają QTc >500 ms i są również w grupie wysokiego ryzyka, β-blokery mają niewystarczającą skuteczność u tych pacjentów, u których zalecono wczesne leczenie ICD (18, 31).
LQTS: Perspektywa Saudyjska
pierwszy raport o LQT z Arabii Saudyjskiej został opublikowany w 1993 roku w szpitalu Sił Zbrojnych w Rijadzie (105). U czworga niemowląt i małych dzieci w wieku od 6 do 48 miesięcy z nawracającymi napadami padaczkowymi w wywiadzie, pochodzących z jednej rodziny, zdiagnozowano LQTS (105). Historia rodziny ujawniła, że dwóch innych członków rodziny miało podobne epizody nagłej utraty przytomności, a trzech członków rodziny zmarło nagle (105). We wszystkich przypadkach wstępną diagnozą była padaczka (105). Kilka lat później odnotowano dwa sporadyczne przypadki ze stosunkowo ostrzejszym wariantem noworodkowych LQT w połączeniu z blokiem AV 2: 1 (106, 107). Wszystkie te opublikowane doniesienia kliniczne nie zawierały żadnych ustaleń genetycznych, które mogłyby wyjaśnić patofizjologię LQTS u nich (105-107).
po raz pierwszy zgłosiliśmy wady genetyczne jako patologiczną podstawę LQTS u podobnych pacjentów z Arabii Saudyjskiej. Zbadaliśmy sześć rodzin z historią omdleń i nagłych niewyjaśnionych zgonów płodu, noworodków i dzieci (12-14 lat). Autosomalny recesywny LQTS1 rozpoznano u dzieci z dwóch rodzin (rycina 3). Autosomalny recesywny LQTS2 rozpoznano w dwóch rodzinach(rycina 4). W jednej rodzinie u pacjentki zdiagnozowano autosomalny dominujący LQTS2 (Fig. 5), u pacjentki występowały napady omdleń w okresie rekonwalescencji poporodowej w szpitalu, co jest bardzo częste u kobiet nosicieli mutacji KCNH2. U wszystkich naszych pacjentów identyfikacja mutacji patogennych w genach kanału jonowego lqts przyczynowego serca doprowadziła do potwierdzonej diagnozy klinicznej, które zostały błędnie zdiagnozowane jako drgawki padaczkowe przed skierowaniem do nas (13, 14) niedawno, Shinwari et al. (15) z King Faisal Specialist Hospital and Research Centre zgłoszono przyczynową mutację lqts1 kcnq1, H258P, w dużej rodzinie z 12 dotkniętymi osobami. Tylko u dwóch nosicieli wystąpiły objawy, ich QTc wynosiło >500 ms, a leki blokujące receptory β-adrenergiczne tłumiły objawy kliniczne u jednego pacjenta, a drugi pacjent z objawem wymagał ICD.
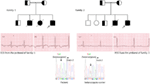
Rysunek 3. Rysunek rodowodowy rodziny-1 i 2 z autosomalnym recesywnym LQTS1, probandy są pokazane strzałką. EKG z probandów w dwóch rodzinach są pokazane w środku, z QTc odpowiednio 557 i 529 ms. U pacjentów stwierdzono mutację introniczną, c.387 -5 T > a w genie KCNQ1 (pokazaną na dole). Mutacja została pokazana za pomocą strzałki. Nie wypełnione koła i kwadraty nie są nośnikami mutacji. Dotknięte osoby są pokazane jako wypełnione koła (żeńskie) i kwadraty (męskie). Pół wypełnione kwadraty i koła to osoby z mutacją heterozygoty. Osoby zmarłe są oznaczone ukośnikami, probandy są oznaczone strzałką, a małżeństwo pokrewieństwa jest wskazane przez = Exon-granica intronu jest pokazana przez kropkowaną linię ze strzałką skierowaną w stronę eksonu.
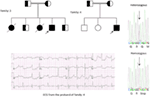
Rysunek 4. Rysunek rodowodowy rodziny – 3 i 4 z autosomalnym recesywnym LQTS2. EKG probanda z rodziny-4 pokazano poniżej rysunku rodowodowego, który pokazuje tachykardię zatokową, prawie kompletny blok AV, szeroki złożony rytm ucieczki z bardzo długimi odstępami QTc (QT >600 ms). Mutacja, c. 3208 C > T (P. Q1070X) w genie KCNH2 została wykryta u pacjentów (pokazana po prawej stronie), oznaczona strzałką.
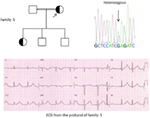
Rysunek 5. U góry po lewej: rodowód rodziny-5. Dotknięty osobnik jest pokazany przez wypełnione koło (kobieta) Proband jest oznaczony strzałką. 12-ołowiowe EKG probanda (I: 2). Badanie przesiewowe genu KCNH2 pokazuje podstawienie nukleotydu “G” na “A” (c.2362G > a, strzałka oznaczona), co prowadzi do podstawienia aminokwasu, P. E788K.
genetyczne i kliniczne odkrycia w naszym badaniu (12-14) z Arabii Saudyjskiej są dość intrygujące z kilku powodów: (1) w sumie zbadaliśmy sześć rodzin, wśród nich cztery były homozygotyczne / złożone heterozygotyczne dla mutacji, a mutacje pochodziły ze źródła przodków; (2) wszystkie mutacje w LQT były nowe, zgłaszane tylko w tych rodzinach Arabskich; (3) ze względu na homozygotyczność lub złożoną heterozygotyczność mutacji, fenotypy kliniczne były również ciężkie w naszych badanych rodzinach (12-14). Sugerowaliśmy, że obserwacje genetyczne i fenotypowe wynikają z ekstremalnie wysokiego wskaźnika małżeństw małżeńskich w Arabii Saudyjskiej (108, 109). Nasze badania dostarczyły pierwszych dowodów naukowych na temat roli pokrewieństwa w odgrywaniu kluczowej roli w niewyjaśnionych arytmiach serca i SCD u dzieci i młodzieży w Arabii Saudyjskiej (12-14 lat). Wykazaliśmy również, że mutacja KCNQ1 c.387 -5 T > a (Nm_000218) (ryc. 3) rozprzestrzeniła się w prowincji Assir w Arabii Saudyjskiej od wspólnego przodka w ciągu kilku pokoleń z powodu wysokiej częstości występowania małżeństw spokrewnionych . Do tej pory jest to najczęstsza mutacja przyczynowa LQTS1 w populacji Arabii Saudyjskiej, która była również obserwowana w Szpitalu Specjalistycznym King Faisal i w szpitalu wojskowym Khamis Mashayt (niepublikowana). Podobny wynik uzyskano również dla mutacji przyczynowej LQTS2, c. 3208 C > T (P.Q1070X) (rycina 4) w genie KCNH2 (12, 14), który jest również mutacją założycielską w Arabii Saudyjskiej i segregowany przez wiele pokoleń w regionie Assir (patrz mapa, rycina 6). Przewidujemy, że istnieje znaczna liczba osób ze wspomnianymi mutacjami przodków/założycieli (zarówno w KCNQ1, jak i KCNH2 i innych genach) w tym regionie, a także w dużych miastach, takich jak Rijad, Dżudda i Dammam, z powodu migracji z miast. W innych prowincjach Arabii Saudyjskiej przewiduje się więcej mutacji założycielskich lub przodków patogennych dla LQT ze względu na wysoki odsetek małżeństw pokrewieństwa.
Rysunek 6. Mapa Arabii Saudyjskiej (dzięki uprzejmości: Wikipedia). Region Assir jest oznaczony grubą kreską, gdzie znaleziono mutacje założycielskie w genach KCNQ1 i kcnh2, opisane w rodzinie 1-4.
ponieważ lqts jest chorobą autosomalną dominującą, spodziewaliśmy się obserwować głównie heterozygotycznych pacjentów przenoszących mutacje. Ale w naszym badaniu zidentyfikowaliśmy głównie pacjentów z mutacjami recesywnymi (12-14). Najwyraźniej ci pacjenci stają się pierwszymi objawami i zostali skierowani głównie do centrum Kardiologicznego księcia sułtana, zwracając na nich naszą uwagę. Niemniej jednak wyniki naszych badań sugerują, że recesywne LQT mogą mieć śmiertelne fenotypy kliniczne u dzieci i nie są rzadkością w Arabii Saudyjskiej (12-14). Ponieważ heterozygotyczni nosiciele mutacji są również podatni na rozwój arytmii i jej powikłań, należy podjąć wspólną inicjatywę, aby przyciągnąć lokalnych lekarzy ogólnych, kardiologów i genetyków klinicznych do wspólnej platformy do identyfikacji osób zagrożonych. Co więcej, w tej chwili nie mamy również informacji genetycznej dla innych rodzinnych zaburzeń arytmii, np. CPVT, SQTS, BrS, Af itp. Wyspecjalizowane ośrodki kardiogenetyczne powinny podjąć inicjatywę poszukiwania wad genetycznych, mutacji i wykonywania badań genotypowo-fenotypowych we wszystkich formach dziedzicznych zaburzeń rytmu serca. Należy również wziąć pod uwagę, że nie wszystkie genetyczne zaburzenia rytmu mają historię rodzinną, ponieważ w wielu przypadkach przyczynowa mutacja arytmii ma pochodzenie de novo, tzn. proband jest pierwszym pacjentem w tej rodzinie z mutacją i jest źródłem przeniesienia mutacji w kolejnych pokoleniach (110, 111). Ze względu na bardzo wysoki odsetek małżeństw małżeńskich w Arabii Saudyjskiej spodziewamy się znacznie większej liczby mutacji założycielskich, które odgrywają kluczową rolę w wrodzonych zaburzeniach rytmu serca w tym kraju. Identyfikacja tych mutacji założycielskich powinna być naszym pierwszym i najważniejszym zadaniem, które ułatwiłoby nam opracowanie skutecznej przedmałżeńskiej, a także przedmałżeńskiej terapii genetycznej w tym kraju. Mutacje w genach przyczynowych LQTS mogą nadawać zmienność penetracji klinicznej, w jednym skrajnym, niektórzy nosiciele mogą być przypuszczalnie całkowicie zdrowi, ale niektórzy nosiciele mogą mieć swój pierwszy przejaw choroby jako omdlenie lub nagła śmierć. Nagła śmierć małego dziecka lub dorosłego stanowi duże obciążenie psychiczne i emocjonalne dla rodziny, badania przesiewowe nosicieli są niezbędne, ponieważ istnieją proste leki, np. β-blokery (również modyfikacja zachowania), które mogą bardzo skutecznie zapobiegać nosicielom śmiertelnych konsekwencji arytmii i SCDs. Osoby z homozygotycznymi mutacjami w genach przyczynowych LQTS mogą mieć ciężką zachorowalność i ekstremalnie wysoki wskaźnik śmiertelności, w tym bradyarytmie płodu, a w wielu przypadkach poronienia, spontaniczne poronienia u ciężarnych matek (12-14).
w Arabii Saudyjskiej nie przeprowadzono zbyt wielu badań na temat występowania różnych SNP w genach związanych z arytmią, a także w genach, które je regulują. Splawski i in. (112) opisał wspólny wariant S1103Y w genie SCN5A związany z arytmią u Afroamerykanów. Allel wariantowy określany jako Y1103 jest odpowiedzialny za przyspieszenie aktywacji kanału, zwiększając tym samym prawdopodobieństwo wystąpienia zaburzeń rytmu serca u osób pochodzenia afrykańskiego (52, 112). K393N jest wariantem genu KCNQ1 zgłaszanym u pacjentów z LQTS1 w USA, ale w populacji Arabskiej wykryliśmy ten wariant U 2% osób (niepublikowane dane). Czy wariant K393N w genie KCNQ1 u Arabów jest porównywalny z wariantem S1103Y (SCN5A) u Afroamerykanów lub wariantem D85N (KCNE1) w populacji japońskiej może również zasługiwać na zbadanie (59).
Oświadczenie o konflikcie interesów
autorzy oświadczają, że badanie zostało przeprowadzone przy braku jakichkolwiek relacji handlowych lub finansowych, które mogłyby być interpretowane jako potencjalny konflikt interesów.
podziękowania
nasza szczera wdzięczność dla Prof. Arthura Wilde ‘ a, Prof. Connie Bezziny i wydawcy czasopisma “Heart” za ich pozwolenie na wykorzystanie rysunku 1 w tym rękopisie z Ref. (113).
1. Bhuiyan ZA. Kliniczne i genetyczne spektrum dziedzicznych zespołów arytmii serca. Amsterdam: Uniwersytet Amsterdamski (2009).
2. Priori SG, Aliot e, Blømstrom-Lundqvist C, Bossaert L, Breithardt G, Brugada P, et al. Task force on sudden cardiac death, European society of cardiology. Europace (2002) 4: 3-18. doi: 10.1053 / eupc.2001.0214
CrossRef Pełny Tekst
3. Wang Q, Shen J, Splawski I, Atkinson D, Li z, Robinson JL, et al. Mutacje SCN5A związane z dziedziczną arytmią serca, zespołem długiego QT. Cell (1995) 80: 805-11. doi:10.1016/0092-8674(95)90359-3
PubMed Streszczenie / PubMed Pełny tekst / CrossRef Pełny tekst
4. Curran ME, Splawski I, Timothy KW, Vincent GM, Green ED, Keating MT. Molekularne podstawy arytmii serca: mutacje HERG powodują zespół długiego QT. Cell (1995) 80: 795-803. doi: 10.1016/0092-8674(95)90358-5
PubMed Streszczenie / PubMed Pełny Tekst / CrossRef Pełny Tekst
5. Wang Q, Curran ME, Splawski I, Burn TC, Millholland JM, VanRaay TJ, et al. Pozycyjne klonowanie nowego genu kanału potasowego: mutacje KVLQT1 powodują zaburzenia rytmu serca. Nat Genet (1996) 12: 17-23. doi: 10.1038 / ng0196-17
PubMed Streszczenie / PubMed Pełny tekst / CrossRef Pełny tekst
6. Splawski I, Tristani-Firouzi M, Lehmann MH, Sanguinetti MC, Keating MT. Mutacje w genie hminK powodują zespół długiego QT i hamują funkcję IKs. Nat Genet (1997) 17:338-40. doi: 10.1038 / ng1197-338
PubMed Abstract | PubMed Full Text / CrossRef Full Text
7. Sanguinetti MC, Jiang C, Curran ME, Keating MT. Mechanistyczny związek między dziedziczną i nabytą arytmią serca: HERG koduje kanał potasowy IKr. Cell (1995) 81: 299-307. doi:10.1016/0092-8674(95)90340-2
PubMed Streszczenie / PubMed Pełny tekst / CrossRef Pełny tekst
8. Neyroud N, Tesson F, Denjoy I, Leibovici m, Donger C, Barhanin J, et al. Nowatorska mutacja w genie kanału potasowego kvlqt1 powoduje zespół Kardioaudytowy Jervella i Lange-Nielsena. Nat Genet (1997) 15:186-9. doi: 10.1038 / ng0297-186
PubMed Streszczenie / PubMed Pełny tekst / CrossRef Pełny tekst
9. Kucera JP, Rohr S, Rudy Y. lokalizacja kanałów sodowych w dyskach interkalowanych moduluje przewodzenie serca. Circ Res (2002) 91: 1176-82. doi: 10.1161/01.RES.0000046237.54156.0 a
PubMed Streszczenie / PubMed Pełny tekst / CrossRef Pełny tekst
10. Oxford EM, Musa H, Maass K, Coombs w, TAFFET SM, Delmar M. Connexin43 remodeling caused by inhibition of plakophilin-2 expression in cardiac cells. Circ Res (2007)101: 703-11. doi: 10.1161 / CIRCRESAHA.107.154252
PubMed Streszczenie / PubMed Pełny Tekst / CrossRef Pełny Tekst
11. Rohr S. molecular crosstalk between mechanical and electrical junctions at the intercalated disc. Circ Res (2007)101: 637-9. doi: 10.1161 / CIRCRESAHA.107.161901
CrossRef Pełny Tekst
12. Bhuiyan ZA, Momenah TS, Gong Q, Amin AS, Ghamdi SA, Carvalho JS, et al. Nawracająca wewnątrzmaciczna utrata płodu spowodowana bliskim brakiem HERG: charakterystyka kliniczna i funkcjonalna homozygotycznej mutacji HERG Q1070X. Heart Rhythm (2008) 5: 553-61. doi: 10.1016 / j.hrthm.2008.01.020
PubMed Abstract / PubMed Full Text / CrossRef Full Text
13. Bhuiyan ZA, Momenah TS, Amin TS, Al-Khadra AS, Alders M, Wilde Aam, et al. Mutacja Introniczna prowadząca do niepełnego pomijania eksonu-2 w KCNQ1 ratuje słuch w zespole Jervella i Lange-Nielsena. Prog Biophys Mol Biol (2008) 98: 319-27. doi: 10.1016 / j.pbiomolbio.2008.10.004
PubMed Abstract / PubMed Full Text / CrossRef Full Text
14. Bhuiyan ZA, Al-Shahrani S, Al-Khadra AS, Al-Ghamdi S, Al-Kalaf K, Mannens MMAM, et al. Analiza kliniczna i genetyczna zespołu długiego QT u dzieci z sześciu rodzin w Arabii Saudyjskiej: czy są różne? Pediatr Cardiol (2009) 30: 490-501. doi: 10.1007 / s00246-008-9377-y
PubMed Abstract / PubMed Full Text / CrossRef Full Text
15. Shinwari ZM, Al-Hazzani a, Dzimiri N, Tulbah S, Mallawi Y, Al-Fayyadh m, et al. Identyfikacja nowej mutacji KCNQ1 w dużej rodzinie z zespołem długiego QT: konsekwencje kliniczne i implikacje prewencyjne. Clin Genet (2013) 83: 370-4. doi: 10.1111 / j.1399-0004.2012.01914.x
PubMed Streszczenie / PubMed Pełny tekst / CrossRef Pełny tekst
16. Schwartz PJ, Stramba-Badiale M, Crotti L, Pedrazzini M, Besana a, Bosi G, et al. Występowanie wrodzonego zespołu długiego QT. (2009) 120:1761-7. doi: 10.1161 / circulation.109.863209
PubMed Streszczenie / PubMed Pełny Tekst / CrossRef Pełny Tekst
17. Schwartz PJ, Moss AJ, Vincent GM, Crampton RS. Kryteria diagnostyczne zespołu długiego QT. Aktualizacja. (1993) 88:782-4. doi: 10.1161/01.CIR.88.2.782
CrossRef Pełny Tekst
18. Priori SG, Wilde AA, Horie M, Cho Y, Behr ER, Berul C, et al. Streszczenie: deklaracja konsensusu HRS/EHRA / APHRS dotycząca diagnostyki i leczenia pacjentów z dziedzicznymi pierwotnymi zespołami arytmii. 15:1389-406. doi: 10.1093/europace / eut272
CrossRef Pełny tekst
19. Liu JF, Jons C, Moss AJ, McNitt S, Peterson DR, Qi m, et al. Rejestr Syndromu. Czynniki ryzyka nawracających omdleń i późniejszych zdarzeń śmiertelnych lub prawie śmiertelnych u dzieci i młodzieży z zespołem wydłużonego odstępu QT. J Am Coll Cardiol (2011) 57: 941-50. doi: 10.1016 / j.jacc.2010.10.025
PubMed Abstract / PubMed Full Text / CrossRef Full Text
20. Splawski I, Shen J, Timothy KW, Lehmann MH, Priori s, Robinson JL, et al. Spektrum mutacji w genach zespołu długiego QT. KVLQT1, HERG, SCN5A, KCNE1 i KCNE2. Circulation (2000)102: 1178-85. doi: 10.1161/01.CIR.102.10.1178
PubMed Streszczenie / PubMed Pełny Tekst / CrossRef Pełny Tekst
21. Westenskow P, Splawski i, Timothy KW, Keating MT, Sanguinetti MC. Mutacje złożone: częstą przyczyną ciężkiego zespołu wydłużonego odstępu QT. (2004) 109:1834-41. doi: 10.1161/01.CIR.0000125524.34234.13
PubMed Abstract | PubMed Full Text / CrossRef Full Text
22. Mullally J, Goldenberg I, Moss AJ, Lopes CM, Ackerman MJ, Zareba W, et al. Ryzyko zagrażających życiu zdarzeń sercowych u pacjentów z zespołem wydłużonego odstępu QT i licznymi mutacjami. 10:378-82 doi: 10.1016 / j.hrthm.2012.11.006
PubMed Abstract / PubMed Full Text / CrossRef Full Text
23. Napolitano C, Priori SG, Schwartz PJ, Bloise R, Ronchetti E, Nastoli J, et al. Badania genetyczne w zespole długiego QT: opracowanie i Walidacja skutecznego podejścia do genotypowania w praktyce klinicznej. JAMA (2005) 294:2975-80. doi: 10.1001 / jama.294.23.2975
PubMed Streszczenie / PubMed Pełny Tekst / CrossRef Pełny Tekst
24. Roden DM. Praktyka kliniczna. Zespół długiego QT. N Engl J Med (2008) 358: 169-76. doi: 10.1056 / NEJMcp0706513
CrossRef Pełny tekst
25. Schwartz PJ, Priori SG, Spazzolini C, Moss AJ, Vincent GM, Napolitano C, et al. Korelacja genotypowo-fenotypowa w zespole długiego QT: specyficzne dla genu czynniki wywołujące zagrażające życiu zaburzenia rytmu serca. Obieg (2001)103: 89-95. doi: 10.1161/01.CIR.103.1.89
CrossRef Pełny Tekst
26. Ackerman MJ, Tester DJ, Porter CJ. Pływanie, specyficzny dla genu arytmogenny czynnik wywołujący dziedziczny zespół długiego QT. Mayo Clin Proc (1999) 74: 1088-94. doi:10.4065/74.11.1088
PubMed Streszczenie / PubMed Pełny tekst / CrossRef Pełny tekst
27. Kapa S, Tester DJ, Salisbury BA, Harris-Kerr C, Pungliya MS, Alders m, et al. Badania genetyczne na zespół długiego QT: odróżnianie mutacji patogennych od łagodnych wariantów. 120:1752-60. doi: 10.1161 / circulation.109.863076
PubMed Streszczenie / PubMed Pełny Tekst / CrossRef Pełny Tekst
28. Moss AJ, Shimizu w, Wilde AA, Towbin JA, Zareba w, Robinson JL, et al. Kliniczne aspekty zespołu długiego QT typu 1 według lokalizacji, typu kodującego i funkcji biofizycznej mutacji obejmujących Gen KCNQ1. (2007) 115:2481-9. doi: 10.1161 / circulation.106.665406
PubMed Streszczenie / PubMed Pełny Tekst / CrossRef Pełny Tekst
29. Amin AS, Giudicessi JR, Tijsen AJ, Spanjaart AM, Reckman YJ, Klemens CA, et al. Warianty w 3 ‘ nieprzetłumaczonym regionie kanału potasowego kodowanego KCNQ1 KV7.1 modyfikują nasilenie choroby u pacjentów z zespołem długiego QT typu 1 w sposób specyficzny dla alleli. Eur Heart J (2012) 33: 714-23. doi: 10.1093/eurheartj / ehr473
PubMed Abstract | PubMed Full Text / CrossRef Full Text
30. Barsheshet a, Goldenberg I, O-Uchi J, Moss AJ, Jons C, Shimizu W, et al. Mutacje w pętlach cytoplazmatycznych kanału KCNQ1 i ryzyko wystąpienia zdarzeń zagrażających życiu: implikacje dla specyficznej dla mutacji odpowiedzi na leczenie β-blokerem w zespole długiego QT typu 1. 1988-96 doi: 10.1161 / circulation.111.048041
PubMed Streszczenie / PubMed Pełny Tekst / CrossRef Pełny Tekst
31. Schwartz PJ, Spazzolini C, Crotti L, Bathen J, Amlie JP, Timothy K, et al. Zespół Jervella i Lange-Nielsena: Historia naturalna, podstawy molekularne i wyniki kliniczne. (2006) 113:783-90. doi: 10.1161 / circulation.105.592899
PubMed Streszczenie / PubMed Pełny Tekst / CrossRef Pełny Tekst
32. Bhuiyan ZA, Wilde AA. IKs w sercu i słuchu, ucho może zrobić z mniej niż serce. Circ Cardiovasc Genet (2013) 6: 141-3. doi: 10.1161 / CIRCGENETICS.113.000143
CrossRef Pełny Tekst
33. Wollnik B, Schroeder BC, Kubisch C, Esperer HD, Wieacker P, Jentsch TJ. Patofizjologiczne mechanizmy dominujących i recesywnych mutacji kanału KVLQT1 K+ stwierdzonych w dziedzicznych zaburzeniach rytmu serca. Hum Mol Genet (1997) 6: 1943-9. doi: 10.1093 / hmg / 6.11.1943
CrossRef Pełny Tekst
34. Wilde AA, Jongbloed RJ, DOEVENDANS PA, Düren DR, Hauer RN, van Langen IM, et al. Bodźce słuchowe jako czynnik wywołujący zdarzenia arytmiczne odróżniają pacjentów związanych z HERG (LQTS2) od pacjentów związanych z KVLQT1 (LQTS1). J Am Coll Cardiol (1999) 33: 327-32. doi: 10.1016 / S0735-1097(98)00578-6
CrossRef Pełny tekst
35. Moss AJ, Zareba w, Kaufman ES, Gartman E, Peterson DR, Benhorin J, et al. Zwiększone ryzyko wystąpienia zdarzeń arytmicznych w zespole wydłużonego odstępu QT z mutacjami w regionie porów kanału potasowego genu ludzkiego ether-a-go-go. Obieg (2002)105: 794-9. doi: 10.1161 / hc0702.105124
PubMed Streszczenie / PubMed Pełny tekst / CrossRef Pełny tekst
36. Lupoglazoff JM, Denjoy I, Villain E, Fressart V, Simon F, Bozio A, et al. Zespół długiego QT u noworodków: zaburzenia przewodzenia związane z mutacjami HERG i bradykardią zatokową z mutacjami KCNQ1. J Am Coll Cardiol (2004) 43: 826-30. doi: 10.1016 / j.jacc.2003.09.049
PubMed Streszczenie / PubMed Pełny Tekst / CrossRef Pełny Tekst
37. Chevalier P, Bellocq C, Millat G, Piqueras E, Potet F, Schott JJ, et al. Torsades de pointes complicating atrioventricular block: evidence for a genetic predyspozycje. Heart Rhythm (2007) 4:170-4. doi: 10.1016 / j.hrthm.2006.10.004
PubMed Abstract / PubMed Full Text / CrossRef Full Text
38. Hoorntje T, Alders M, van tintelen p, van der Lip K, Sreeram N, van der Wal A, et al. Homozygotyczne przedwczesne obcinanie białka HERG: ludzki knockout HERG. (1999) 100:1264-7. doi: 10.1161/01.CIR.100.12.1264
PubMed Streszczenie / PubMed Pełny Tekst / CrossRef Pełny Tekst
39. Piippo K, Laitinen P, Swan H, Toivonen L, Viitasalo M, Pasternack m, et al. Homozygotyzm mutacji kanału potasowego HERG powoduje ciężką postać zespołu długiego QT: identyfikację pozornej mutacji założycielskiej u Finów. J Am Coll Cardiol (2000) 35: 1919-25. doi: 10.1016 / S0735-1097(00)00636-7
PubMed Streszczenie / PubMed Pełny tekst / CrossRef Pełny tekst
40. Johnson Wh Jr, Yang P, Yang T, Lau YR, Mostella BA, Wolff DJ, et al. Kliniczna, genetyczna i biofizyczna charakterystyka homozygotycznej mutacji HERG powodującej ciężki noworodkowy zespół długiego QT. Pediatr Res (2003) 53: 744-8. doi: 10.1203/01.PDR.0000059750.17002.B6
PubMed Streszczenie / PubMed Pełny Tekst / CrossRef Pełny Tekst
41. Ackerman MJ, Tester DJ, Jones GS, Will ML, Burrow CR, Curran ME. Różnice etniczne w wariantach kanału potasowego serca: implikacje dla genetycznej podatności na nagłą śmierć sercową i badania genetyczne dla wrodzonego zespołu długiego QT. Mayo Clin Proc (2003) 78: 1479-87. doi:10.4065/78.12.1479
PubMed Streszczenie / PubMed Pełny tekst / CrossRef Pełny tekst
42. Crotti L, Lundquist AL, Insolia R, Pedrazzini m, Ferrandi C, De Ferrari GM, et al. KCNH2-K897T jest genetycznym modyfikatorem utajonego wrodzonego zespołu długiego QT. Obieg (2005)112: 1251-8. doi: 10.1161 / circulation.105.549071
PubMed Streszczenie / PubMed Pełny Tekst / CrossRef Pełny Tekst
43. Nof E, Cordeiro JM, Pérez GJ, Scornik FS, Calloe K, Love B, et al. Wspólny polimorfizm pojedynczego nukleotydu może zaostrzyć zespół długiego QT typu 2 prowadzący do nagłej śmierci niemowlęcia. Circ Cardiovasc Genet (2010) 3:199-206. doi: 10.1161 / CIRCGENETICS.109.898569
PubMed Streszczenie / PubMed Pełny Tekst / CrossRef Pełny Tekst
44. Bezzina C, Veldkamp MW, van Den Berg MP, Postma AV, Rook MB, Viersma JW, et al. Pojedyncza mutacja kanału Na (+) powodująca zarówno zespoły long-QT, jak i Brugada. Circ Res (1999) 85: 1206-13. doi: 10.1161/01.OZE.85.12.1206
PubMed Streszczenie / PubMed Pełny tekst / CrossRef Pełny tekst
45. Kyndt F, Probst V, Potet F, Demolombe S, Chevallier JC, Baro I, et al. Nowa mutacja SCN5A prowadząca do izolowanej wady przewodzenia serca lub zespołu Brugada w dużej rodzinie francuskiej. Obieg (2001)104: 3081-6. doi: 10.1161 / hc5001.100834
PubMed Streszczenie / PubMed Pełny tekst / CrossRef Pełny tekst
46. Makita N, Behr E, Shimizu W, Horie M, Sunami a, Crotti L, et al. Mutacja E1784K w SCN5A jest związana z mieszanym fenotypem klinicznym zespołu długiego QT typu 3. J Clin Invest (2008) 118:2219-29. doi: 10.1172 / JCI34057
PubMed Abstract | PubMed Full Text / CrossRef Full Text
47. Keller DI, Acharfi S, Delacrétaz E, Benammar N, Rotter m, Pfammatter JP, et al. Nowa mutacja w SCN5A, delQKP 1507-1509, powodująca zespół długiego QT: rola pozostałości Q1507 w inaktywacji kanału sodowego. J Mol Cell Cardiol (2003) 35: 1513-21. doi: 10.1016 / j.yjmcc.2003.08.007
PubMed Streszczenie / PubMed Pełny Tekst / CrossRef Pełny Tekst
48. Priori SG, Schwartz PJ, Napolitano C, Bloise R, Ronchetti E, Grillo m, et al. Stratyfikacja ryzyka w zespole długiego QT. N Engl J Med (2003) 348: 1866-74. doi: 10.1056 / NEJMoa022147
PubMed Abstract | PubMed Full Text / CrossRef Full Text
49. Lupoglazoff JM, Cheav T, Baroudi G, Berthet m, Denjoy I, Cauchemez B, et al. Homozygotyczna mutacja SCN5A w zespole długiego QT z funkcjonalnym blokiem przedsionkowo-komorowym 2 do 1. Circ Res (2001) 89: E16–21. doi: 10.1161 / hh1401.095087
CrossRef Pełny tekst
50. Shi R, Zhang y, Yang C, Huang C, Zhou X, Qiang h, et al. Mutacja kanału sodowego serca delQKP 1507-1509 jest związana z rozszerzającym się spektrum fenotypowym LQT3, zaburzeniem przewodzenia, kardiomiopatią rozstrzeniową i wysoką częstością nagłej śmierci młodzieży. Europace (2008)10: 1329-35. doi: 10.1093/europace / eun202
PubMed Abstract | PubMed Full Text / CrossRef Full Text
51. Zumhagen S, Veldkamp MW, Stallmeyer B, Baartscheer a, Eckardt L, Paul M, et al. Heterozygotyczna delecja w genie kanału sodowego serca SCN5A z cechami utraty i wzmocnienia funkcji objawia się jako izolowana choroba przewodzenia, bez objawów Brugady lub zespołu długiego QT. PLoS One (2013) 8:e67963. doi: 10.1371 / dziennik.pone.0067963
PubMed Streszczenie / PubMed Pełny Tekst / CrossRef Pełny Tekst
52. Roślina LD, Bowers PN, Liu Q, Morgan T, Zhang T, State MW, et al. A common cardiac sodium channel variant associated with sudden infant death in African Americans, SCN5A S1103Y. J Clin Invest (2006) 116:430-5. doi: 10.1172 / JCI25618
PubMed Streszczenie / PubMed Pełny tekst / CrossRef Pełny tekst
53. Mohler PJ, Schott JJ, Gramolini ao, Dilly KW, Guatimosim S, duBell WH, et al. Mutacja Ankyrin-B powoduje zaburzenia rytmu serca typu 4 o długim odstępie QT i nagłą śmierć sercową. Nature (2003) 421: 634-9. doi: 10.1038 / nature01335
PubMed Abstract | PubMed Full Text / CrossRef Full Text
54. Schott JJ, Charpentier F, Peltier S, Foley P, Drouin e, Bouhour JB, et al. Mapowanie genu zespołu długiego QT na chromosom 4q25-27. Am J Hum Genet (1995) 57: 1114-22.
PubMed Streszczenie / PubMed Pełny Tekst
55. Białka Barhanin J, Lesage F, Guillemare E, Fink M, Lazdunski M, Romey G. K(V)LQT1 i LSK (minK) łączą się tworząc potasowy prąd sercowy I(Ks). Nature (1996) 384: 78-80. doi: 10.1038 / 384078a0
PubMed Streszczenie / PubMed Pełny tekst / CrossRef Pełny tekst
56. Sanguinetti MC, Curran ME, Zou a, Shen J, Spector PS, Atkinson DL, et al. Współwystępowanie białek K (V)LQT1 i norek(ISK) w celu utworzenia kanału potasowego serca I (Ks). Nature (1996) 384: 80-3. doi: 10.1038 / 384080a0
PubMed Streszczenie / PubMed Pełny tekst / CrossRef Pełny tekst
57. Schulze-Bahr E, Wang Q, Wedekind H, Haverkamp w, Chen Q, Sun Y, et al. Mutacje KCNE1 powodują zespół Jervella i Lange-Nielsena. Nat Genet (1997) 17:267-8. doi: 10.1038 / ng1197-267
CrossRef Pełny tekst
58. Duggal P, Vesely MR, Wattanasirichaigoon D, Villafane J, Kaushik V, Beggs AH. Mutacja genu IKs związana zarówno z formami Jervella i Lange-Nielsena, jak i Romano-Warda zespołu długiego QT. (1998) 97:142-6. doi: 10.1186/1471-2350-9-24
PubMed Streszczenie / PubMed Pełny Tekst / CrossRef Pełny Tekst
59. Nishio y, Makiyama T, Itoh H, Sakaguchi T, Ohno S, Gong YZ i in. D85N, polimorfizm KCNE1, jest chorobotwórczym wariantem genu w zespole długiego QT. J Am Coll Cardiol (2009) 54: 812-9. doi: 10.1016 / j.jacc.2009.06.005
PubMed Abstract / PubMed Full Text / CrossRef Full Text
60. Paulussen AD, Gilissen RA, Armstrong m, DOEVENDANS PA, Verhasselt P, Smeets HJ, et al. Zmiany genetyczne KCNQ1, KCNH2, SCN5A, KCNE1 i kcne2 u pacjentów z indukowanym przez leki zespołem długiego QT. Mol Med (2004) 82: 182-8. doi: 10.1007 / s00109-003-0522-z
PubMed Abstract / PubMed Full Text / CrossRef Full Text
61. Abbott GW, Sesti F, Splawski I, Buck ME, Lehmann MH, Timothy KW, et al. MiRP1 tworzy kanały potasowe IKr z HERG i jest związany z arytmią serca. Cell (1999) 97: 175-87. doi: 10.1016/S0092-8674(00)80728-X
PubMed Streszczenie / PubMed Pełny tekst / CrossRef Pełny tekst
62. Gordon e, Panaghie G, Deng L, Bee KJ, Roepke TK, Krogh-Madsen T, et al. Mutacja KCNE2 u pacjenta z arytmią serca wywołaną bodźcami słuchowymi i zaburzeniami elektrolitowymi w surowicy. Cardiovasc Res (2008) 77: 98-106. doi: 10.1093/cvr / cvm030
PubMed Streszczenie / PubMed Pełny tekst / CrossRef Pełny tekst
63. Tynk NM, Tawil R, Tristani-Firouzi M, Canún S, Bendahhou S, Tsunoda a i in. Mutacje w Kir2.1 powodują rozwojowe i epizodyczne fenotypy elektryczne zespołu Andersena. Cell (2001)105: 511-9. doi: 10.1016 / S0092-8674(01)00342-7
PubMed Streszczenie / PubMed Pełny Tekst / CrossRef Pełny Tekst
64. Kubo Y, Baldwin TJ, Jan YN, Jan LY. Struktura pierwotna i ekspresja funkcjonalna kanału prostowniczego mysi. Nature (1993) 362: 127-33. doi: 10.1038 / 362127a0
PubMed Abstract | PubMed Full Text / CrossRef Full Text
65. Raab-Graham KF, Radeke CM, Vandenberg CA. Klonowanie molekularne i ekspresja ludzkiego serca wewnątrz prostownika kanału potasowego. Neuroreport (1994) 5: 2501-5. doi: 10.1097/00001756-199412000-00024
PubMed Streszczenie / PubMed Pełny Tekst / CrossRef Pełny Tekst
66. Splawski i, Timothy KW, Sharpe LM, Decher N, Kumar P, Bloise R, et al. Ca (V) 1.2 dysfunkcja kanału wapniowego powoduje zaburzenia wielosystemowe, w tym arytmię i autyzm. Cell (2004)119: 19-31. doi: 10.1016 / j.cell.2004.09.011
PubMed Abstract / PubMed Full Text / CrossRef Full Text
67. Splawski i, Timothy KW, Decher N, Kumar P, Sachse FB, Beggs AH, et al. Ciężkie zaburzenia rytmu serca spowodowane przez mutacje kanału wapniowego typu L serca. Proc Natl Acad Sci U S A (2005)102: 8089-96. doi: 10.1073 / pnas.0502506102
PubMed Abstract / PubMed Full Text / CrossRef Full Text
68. Gillis J, Burashnikov E, Antzelevitch C, Blaser S, Gross G, Turner L, et al. Długie QT, syndaktylia, przykurcze stawów, udar mózgu i nowa mutacja CACNA1C: rozszerzenie spektrum zespołu Timothy ‘ ego. Am J Med Genet A (2012) 158A:182-7. doi: 10.1002 / ajmg.a.34355
PubMed Streszczenie / PubMed Pełny Tekst / CrossRef Pełny Tekst
69. Dufendach KA, Giudicessi JR, Boczek NJ, Ackerman MJ. Mozaicyzm matczyny utrudnia rozpoznanie noworodka zespołu Tymoteusza typu 1. Pediatria (2013) 131:e1991-5. doi: 10.1542 / peds.2012-2941
PubMed Streszczenie / PubMed Pełny Tekst / CrossRef Pełny Tekst
70. Vatta M, Ackerman MJ, Ye B, Makielski JC, Ughanze EE, Taylor EW, et al. Zmutowana caveolina-3 indukuje uporczywy późny prąd sodowy i jest związana z zespołem długiego QT. Obieg (2006) 114:2104–12. doi: 10.1161 / circulation.106.635268
PubMed Streszczenie / PubMed Pełny Tekst / CrossRef Pełny Tekst
71. Cronk LB, Ye B, Kaku T, Tester DJ, Vatta M, Makielski JC i in. Nowatorski mechanizm zespołu nagłej śmierci niemowląt: uporczywy późny prąd sodowy wtórny do mutacji caveolin-3. Heart Rhythm (2007) 4:161-6. doi: 10.1016 / j.hrthm.2006.11.030
PubMed Abstract / PubMed Full Text / CrossRef Full Text
72. Engelman ja, Zhang X, Galbiati F, Volonte D, Sotgia F, Pestell RG, et al. Genetyka molekularna z rodziny genów caveolin: implikacje dla ludzkich nowotworów, cukrzycy, choroby Alzheimera i dystrofii mięśniowej. Am J Hum Genet (1998) 63: 1578-87. doi:10.1086/302172
CrossRef Pełny tekst
73. Balijepalli RC, Foell JD, Hall DD, Hell JW, Kamp TJ. Lokalizacja kanałów serca typu L Ca (2+) do kompleksu sygnałowego caveolar macromolecular jest wymagana do regulacji beta (2) – adrenergicznej. Proc Natl Acad Sci U S A (2006) 103:7500-5. doi: 10.1073 / pnas.0503465103
PubMed Abstract / PubMed Full Text / CrossRef Full Text
74. Medeiros-Domingo a, Kaku T, Tester DJ, Iturralde-Torres P, Itty a, Ye B, et al. SCN4B-kodowana podjednostka kanału sodowego beta4 w wrodzonym zespole długiego QT. (2007) 116:134-42. doi: 10.1161 / circulation.106.659086
PubMed Streszczenie / PubMed Pełny Tekst / CrossRef Pełny Tekst
75. Chen L, Marquardt ML, Tester DJ, Sampson KJ, Ackerman MJ, Kass RS. Mutacja białka zakotwiczającego kinazę A powoduje zespół długiego QT. Proc Natl Acad Sci U S A (2007) 104:20990-5. doi: 10.1073 / pnas.0710527105
PubMed Abstract / PubMed Full Text / CrossRef Full Text
76. Wu g, AI T, Kim JJ, Mohapatra B, Xi Y, Li Z i in. Mutacja alfa-1-syntrofiny i zespół długiego QT: choroba zaburzeń kanału sodowego. Circ Arrhythm Electrophysiol (2008) 1:193-201. doi: 10.1161 / CIRCEP.108.769224
PubMed Streszczenie / PubMed Pełny Tekst / CrossRef Pełny Tekst
77. Yang y, Yang y, Liang B, Liu J, Li J, Grunnet m, et al. Identyfikacja Kir3.4 mutacja w wrodzonym zespole długiego QT. Am J Hum Genet (2010) 86: 872-80. doi: 10.1016 / j.ajhg.2010.04.017
PubMed Streszczenie / PubMed Pełny Tekst / CrossRef Pełny Tekst
78. Roden DM. Mechanizmy i postępowanie w proarytmii. Am J Cardiol (1998) 82:49i–57I. doi:10.1016/S0002-9149(98)00472-X
CrossRef Pełny tekst
79. Mitcheson JS, Chen J, Lin m, Culberson C, Sanguinetti MC. Strukturalna podstawa zespołu długiego QT wywołanego lekami. Proc Natl Acad Sci U S A (2000) 97: 12329-33. doi: 10.1073 / pnas.210244497
CrossRef Pełny Tekst
80. Kannankeril PJ, Roden DM. Drug-induced long QT and torsade de pointes: recent advances. Curr Opin Cardiol (2007)22: 39-43. doi: 10.1097 / HCO.0b013e32801129eb
PubMed Streszczenie / PubMed Pełny tekst / CrossRef Pełny tekst
81. Veerman CC, Verkerk AO, Blom MT, Klemens CA, Langendijk PN, van Ginneken AC, et al. Powolne opóźnione blokowanie prądu potasowego prostownika przyczynia się w istotny sposób do wywołanego przez leki zespołu długiego QT. Circ Arrhythm Electrophysiol (2013) 6: 1002-9. doi: 10.1161 / CIRCEP.113.000239
PubMed Streszczenie / PubMed Pełny Tekst / CrossRef Pełny Tekst
82. Sesti F, Abbott GW, Wei J, Murray KT, Saksena S, Schwartz PJ, et al. Częsty polimorfizm związany z arytmią serca wywołaną antybiotykami. Proc Natl Acad Sci U S A (2000) 97: 10613-8. doi: 10.1073 / pnas.180223197
PubMed Streszczenie / PubMed Pełny Tekst / CrossRef Pełny Tekst
83. Kääb S, Crawford DC, Sinner MF, Behr ER, Kannankeril PJ, Wilde AA, et al. Duże badanie genów kandydujących identyfikuje polimorfizm KCNE1 D85N jako możliwy modulator torsades de pointes indukowanych lekami. Circ Cardiovasc Genet (2012) 5: 91-9. doi: 10.1161 / CIRCGENETICS.111.960930
PubMed Streszczenie / PubMed Pełny Tekst / CrossRef Pełny Tekst
84. Nakamura K, Katayama y, Kusano KF, Haraoka K, Tani y, Nagase S, et al. Zespół długiego QT indukowany przeciwciałami anty-KCNH2: Nowa nabyta postać zespołu długiego QT. J Am Coll Cardiol (2007) 50: 1808-9. doi: 10.1016 / j.jacc.2007.07.037
CrossRef Pełny Tekst
85. Moss AJ, Zareba w, Benhorin J, Locati EH, Hall WJ, Robinson JL, et al. EKG wzorce fali T w genetycznie odmiennych formach dziedzicznego zespołu długiego QT. (1995) 92:2929-34. doi: 10.1161/01.CIR.92.10.2929
PubMed Streszczenie / PubMed Pełny Tekst / CrossRef Pełny Tekst
86. Zhang L, Timothy KW, Vincent GM, Lehmann MH, Fox J, Giuli LC, et al. Spektrum wzorców fal ST-T i parametrów repolaryzacji w wrodzonym zespole długiego QT: wyniki EKG identyfikują genotypy. Obieg (2000) 102:2849-55. doi: 10.1161/01.CIR.102.23.2849
PubMed Streszczenie / PubMed Pełny Tekst / CrossRef Pełny Tekst
87. Tan HL, Bardai a, Shimizu w, Moss AJ, Schulze-Bahr E, Noda T, et al. Wystąpienie arytmii w wrodzonym zespole wydłużonego odstępu QT specyficzne dla genotypu: możliwe implikacje terapeutyczne. (2006) 114:2096-103. doi: 10.1161 / circulation.106.642694
PubMed Streszczenie / PubMed Pełny Tekst / CrossRef Pełny Tekst
88. Locati EH, Zareba w, Moss AJ, Schwartz PJ, Vincent GM, Lehmann MH, et al. Różnice związane z wiekiem i płcią w objawach klinicznych u pacjentów z wrodzonym zespołem wydłużonego odstępu QT: wyniki Międzynarodowego rejestru LQTS. Obieg (1998) 97: 2237-44. doi: 10.1161/01.CIR.97.22.2237
PubMed Streszczenie / PubMed Pełny Tekst / CrossRef Pełny Tekst
89. Zareba w, Moss AJ, Schwartz PJ, Vincent GM, Robinson JL, Priori SG, et al. Wpływ genotypu na przebieg kliniczny zespołu wydłużonego odstępu QT. International Long-QT Syndrome Registry Research Group. N Engl J Med (1998) 339: 960-5. doi: 10.1056 / NEJM199810013391404
PubMed Streszczenie / PubMed Pełny tekst / CrossRef Pełny tekst
90. Goldenberg I, Moss AJ, Bradley J, Polonsky s, Peterson DR, McNitt s, et al. Zespół wydłużonego QT po 40. roku życia. (2008) 117:2192-201. doi: 10.1161 / circulation.107.729368
PubMed Streszczenie / PubMed Pełny Tekst / CrossRef Pełny Tekst
91. Zareba w, Moss AJ, Locati EH, Lehmann MH, Peterson DR, Hall WJ, et al. Rejestr Syndromu. Modulujący wpływ wieku i płci na przebieg kliniczny zespołu wydłużonego odstępu QT w zależności od genotypu. J Am Coll Cardiol (2003) 42: 103-9. doi: 10.1016 / S0735-1097(03)00554-0
PubMed Streszczenie / PubMed Pełny tekst / CrossRef Pełny tekst
92. Seth R, Moss AJ, McNitt s, Zareba w, Andrews ML, Qi m, et al. Zespół długiego QT a ciąża. J Am Coll Cardiol (2007) 49: 1092-8. doi: 10.1016 / j.jacc.2006.09.054
PubMed Abstract / PubMed Full Text / CrossRef Full Text
93. Schwartz PJ, Priori SG, Dumaine R,Napolitano C, Antzelevitch C, Stramba-Badiale m, et al. Molekularny związek między zespołem nagłej śmierci niemowląt a zespołem długiego QT. N Engl J Med (2000) 343: 262-7. doi: 10.1056 / NEJM200007273430405
CrossRef Pełny tekst
94. Schwartz PJ, Priori SG, Bloise R, Napolitano C, Ronchetti E, Piccinini A, et al. Diagnostyka molekularna u dziecka z zespołem nagłej śmierci niemowląt. Lancet (2001) 358: 1342-3. doi: 10.1016 / S0140-6736(01)06450-9
PubMed Streszczenie / PubMed Pełny tekst / CrossRef Pełny tekst
95. Christiansen M, Tønder N, Larsen LA, Andersen PS, Simonsen H, Oyen N, et al. Mutacje w kanale hERG K+-ion: nowatorski związek między zespołem długiego QT a zespołem nagłej śmierci niemowląt. Am J Cardiol (2005) 95: 433-4. doi: 10.1016 / j.amjcard.2004.09.054
PubMed Streszczenie / PubMed Pełny Tekst / CrossRef Pełny Tekst
96. Tester DJ, Ackerman MJ. Pośmiertny zespół wydłużonego QT badania genetyczne na nagłą niewyjaśnioną śmierć młodych. J Am Coll Cardiol (2007) 49: 240-6. doi: 10.1016 / j.jacc.2006.10.010
PubMed Abstract / PubMed Full Text / CrossRef Full Text
97. Hofman N, Tan HL, Clur SA, Alders m, van Langen IM, Wilde AA. Udział dziedzicznej choroby serca w nagłej śmierci sercowej w dzieciństwie. Pediatria (2007) 120:e967-73. doi: 10.1542 / peds.2006-3751
PubMed Streszczenie / PubMed Pełny Tekst / CrossRef Pełny Tekst
98. Wedekind H, Smits JP, Schulze-Bahr E, Arnold R, Veldkamp MW, Bajanowski T, et al. Mutacja De novo w genie SCN5A związana z wczesnym początkiem nagłej śmierci niemowlęcia. Obieg (2001) 104:1158-64. doi: 10.1161 / hc3501.095361
CrossRef Pełny tekst
99. Wang DW, Desai RR, Crotti L, Arnestad M, Insolia R, Pedrazzini m, et al. Dysfunkcja kanału sodowego serca w zespole nagłej śmierci niemowląt. (2007) 115: 368-76. doi: 10.1161 / circulation.106.646513
PubMed Streszczenie / PubMed Pełny Tekst / CrossRef Pełny Tekst
100. Van Norstrand DW, Valdivia CR, Tester DJ, Ueda K, London B, Makielski JC, et al. Molekularna i funkcjonalna charakterystyka nowych mutacji genu podobnego do dehydrogenazy glicerolu-3-fosforanu 1 (GPD1-L) w zespole nagłej śmierci niemowląt. (2007) 116:2253-9. doi: 10.1161 / obieg.107.704627
PubMed Streszczenie / PubMed Pełny Tekst / CrossRef Pełny Tekst
101. Tan BH, Pundi KN, Van Norstrand DW, Valdivia CR, Tester DJ, Medeiros-Domingo a, et al. Zespół nagłej śmierci niemowląt – związane z mutacjami w podjednostkach kanału sodowego beta. Heart Rhythm (2010) 7: 771-8. doi: 10.1016 / j.hrthm.2010.01.032
PubMed Abstract / PubMed Full Text / CrossRef Full Text
102. Miller TE, Estrella E, Myerburg RJ, Garcia de Viera J, Moreno N, Rusconi P, et al. Nawracająca utrata płodu w trzecim trymestrze ciąży i mozaicyzm matki w zespole długiego QT. Obieg (2004)109: 3029-34. doi: 10.1161/01.CIR.0000130666.81539.9 E
PubMed Streszczenie / PubMed Pełny Tekst / CrossRef Pełny Tekst
103. Chockalingam P, Crotti L, Girardengo G, Johnson JN, Harris KM, van der Heijden JF, et al. Nie wszystkie beta-blokery są równe w leczeniu zespołu długiego QT typu 1 i 2: wyższe nawroty zdarzeń pod metoprololem. J Am Coll Cardiol (2012) 60: 2092-6. doi: 10.1016 / j.jacc.2012.07.046
PubMed Streszczenie / PubMed Pełny Tekst / CrossRef Pełny Tekst
104. Schwartz PJ, Priori SG, Cerrone M, Spazzolini C, Odero a, Napolitano C, et al. Osłabienie współczulne lewego serca w leczeniu pacjentów wysokiego ryzyka dotkniętych zespołem wydłużonego odstępu QT. (2004) 109:1826-33. doi: 10.1161/01.CIR.0000125523.14403.1 E
PubMed Streszczenie / PubMed Pełny Tekst / CrossRef Pełny Tekst
105. Singh B, al Shahwan SA, Habbab MA, al Deeb SM, Biary N. idiopatyczny zespół długiego QT: zadawanie właściwego pytania. Lancet (1993) 341:741. doi:10.1016/0140-6736(93)90501-7
CrossRef Pełny tekst
106. Gorgels AP, Al Fadley F, Zaman L, Kantoch MJ, Al Halees Z. The long QT syndrome with disabled atrioventricular conduction: a malignant variant in infants. J Cardiovasc Electrophysiol (1998) 9: 1225-32. doi: 10.1111 / j.1540-8167.1998.tb00096x
PubMed Streszczenie / PubMed Pełny tekst / CrossRef Pełny tekst
107. Kantoch MJ, Qurashi MM, Bulbul ZR, Gorgels AP. Noworodek ze złożoną wrodzoną chorobą serca, blokiem przedsionkowo-komorowym i częstoskurczem komorowym torsade de pointes. Pacing Clin Electrophysiol (1998) 21:2664-7. doi: 10.1111 / j.1540-8159.1998.tb00043X
CrossRef Pełny tekst
108. El-Hazmi MA, al-Swailem AR, Warsy AS, al-Swailem AM, Sulaimani R, al-Meshari AA. Pokrewieństwo wśród ludności Arabii Saudyjskiej. J Med Genet (1995) 32: 623-6. doi: 10.1136 / jmg.32.8.623
CrossRef Pełny Tekst
109. El Mouzan MI, Al Salloum AA, Al Herbish AS, Qurachi MM, Al Omar AA. Consanguinity and major genetic disorders in Saudi children: a community-based cross-sectional study. Ann Saudi Med (2008)28: 169-73. doi:10.4103/0256-4947.51726
PubMed Streszczenie / PubMed Pełny tekst / CrossRef Pełny tekst
110. Medeiros-Domingo a, Bhuiyan ZA, Tester DJ, Hofman N, Bikker H, van tintelen JP, et al. Kodowany RYR2 kanał uwalniania receptora ryanodyny / wapnia u pacjentów, u których wcześniej zdiagnozowano polimorficzny tachykardię komorową katecholaminergiczną lub zespół wydłużonego odstępu QT wywołany wysiłkiem fizycznym: kompleksowa Otwarta analiza mutacyjna ramek odczytu. J Am Coll Cardiol (2009) 54: 2065-74. doi: 10.1016 / j.jacc.2009.08.022
PubMed Abstract / PubMed Full Text / CrossRef Full Text
111. Al-Aama JY, Al-Ghamdi S, Bdier AY, Wilde AA, Bhuiyan ZA. De novo mutacja w genie KCNQ1 przyczynowa do zespołu Jervella i Lange-Nielsena. Clin Genet (2013). doi: 10.1111 / cge.12300
PubMed Streszczenie / PubMed Pełny Tekst / CrossRef Pełny Tekst
112. Splawski i, Timothy KW, Tateyama M, Clancy CE, Malhotra a, Beggs AH, et al. Wariant kanału sodowego SCN5A związany z ryzykiem arytmii serca. Nauka (2002) 297:1333-6. 10.1126 / nauka1073569
CrossRef Pełny Tekst
113. Wilde AA, Bezzina CR. Genetyka zaburzeń rytmu serca. Heart (2005) 91:1352-8.