Genetyka rozszczepu wargi i podniebienia
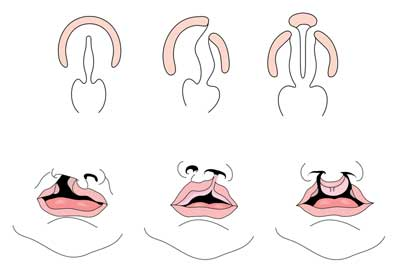 Intro / abstractCleft warga z rozszczepem podniebienia lub bez niego jest złożoną wadą wrodzoną, którą można wyizolować lub zobaczyć razem z innymi wadami rozwojowymi. Może być również częścią fenotypu zespołu genetycznego. Ten artykuł służy jako przegląd częstości występowania rozszczepu wargi i podniebienia, ryzyka nawrotu i ryzyka innych wad wrodzonych. Zbadane zostaną zespoły genetyczne i narażenie teratogenne związane z rozszczepami jamy ustnej. Ponadto omówione zostaną badania genetyczne powszechnie wymagane w pediatrycznej genetyce klinicznej w celu oceny pacjenta z rozszczepem wargi i podniebienia.
Intro / abstractCleft warga z rozszczepem podniebienia lub bez niego jest złożoną wadą wrodzoną, którą można wyizolować lub zobaczyć razem z innymi wadami rozwojowymi. Może być również częścią fenotypu zespołu genetycznego. Ten artykuł służy jako przegląd częstości występowania rozszczepu wargi i podniebienia, ryzyka nawrotu i ryzyka innych wad wrodzonych. Zbadane zostaną zespoły genetyczne i narażenie teratogenne związane z rozszczepami jamy ustnej. Ponadto omówione zostaną badania genetyczne powszechnie wymagane w pediatrycznej genetyce klinicznej w celu oceny pacjenta z rozszczepem wargi i podniebienia.
rozszczep wargi z lub bez rozszczepu podniebienia (CL/CP) różni się od izolowanego rozszczepu podniebienia (CP) na poziomie embrionalnym, epidemiologicznym i genetycznym. Rozszczep wargi zazwyczaj wynika z wyeksponowania szczęki i wyeksponowania przyśrodkowego nosa, które nie łączą się między piątym a szóstym tygodniem rozwoju embrionalnego. Prawidłowy rozwój podniebienia wynika z powstawania podniebienia pierwotnego i podniebienia wtórnego. Pierwotne podniebienie powstaje w tygodniu od szóstego do siódmego przez rozwój i fuzję procesów przyśrodkowych nosa, bocznych nosa i szczęki. Podniebienie wtórne pochodzi z półek podniebiennych (które rozwijają się w wyniku sparowanych procesów szczękowych pierwszego łuku rozgałęziowego), stając się poziome i zespolone, tworząc podniebienie twarde i miękkie około dziewiątego tygodnia rozwoju embrionalnego. Półki łączą się również z przegrodą nosową i podniebieniem pierwotnym. (1)
rozszczepy jamy ustnej są jedną z najczęstszych wad wrodzonych obserwowanych w przedszkolu noworodków, z ogólną częstością 1.6 na tysiąc noworodków na całym świecie, przy CL/CP obserwowanym w około jednym na tysiąc urodzeń i CP obserwowanym w 0,6 na tysiąc urodzeń. (2) istnieje wyższa częstotliwość CL/CP u osób pochodzenia azjatyckiego, afrykańskiego i indiańskiego. CL / CP jest również bardziej powszechne u mężczyzn. W przeciwieństwie do tego, nie ma znaczącej różnicy w częstości występowania CP wśród różnych środowisk etnicznych, a CP jest bardziej powszechne u kobiet. (3) ryzyko nawrotu w rodzinie zależy od tego, czy rozszczep jest izolowany (bez innych wyników klinicznych) lub postrzegany jako część zespołu genetycznego. Większość przypadków rozszczepów jamy ustnej jest izolowana (około 80%). Uważa się, że pojedyncze rozszczepy mają dziedziczenie wieloczynnikowe: są one spowodowane kombinacją wielu czynników, zarówno genetycznych, jak i środowiskowych. Ryzyko nawrotu (Tabela 1) zwiększa się, gdy występuje więcej niż jeden krewny dotknięty chorobą. Ryzyko nawrotu również zwiększa się, im poważniejsza jest wada.

rozszczep wargi i podniebienia można zobaczyć z innymi wadami wrodzonymi. Prawdopodobieństwo etiologii genetycznej lub teratogennej zwiększa się, im więcej wad wrodzonych, z którymi pacjent się prezentuje. Obecność innych problemów, takich jak niepełnosprawność intelektualna, problemy behawioralne, takie jak autyzm, cechy dysmorficzne lub inne problemy medyczne, również sprawi, że zaburzenie genetyczne lub ekspozycja teratogenna będą bardziej prawdopodobne. Około 13% osób z rozszczepem wargi będzie miało inne problemy medyczne lub anomalie. Liczba ta wzrasta do 37% z rozszczepem wargi i podniebienia oraz do 47% z rozszczepem podniebienia samego.
wykazano, że prenatalna ekspozycja na czynniki teratogenne (takie jak talidomid, leki przeciwdrgawkowe, alkohol, kwas retinowy i papierosy) i choroby matki (takie jak cukrzyca, różyczka i niedobór kwasu foliowego) zwiększają ryzyko rozszczepów jamy ustnej. Obecność pasm owodniowych zwiększa również ryzyko rozszczepów. Wiadomo, że suplementacja kwasu foliowego zmniejsza ryzyko rozszczepów jamy ustnej.
Sekwencja Pierre ‘ a Robina jest anomalią czaszkowo-twarzową charakteryzującą się hipoplazją żuchwy lub mikrognatią, wtórnym rozszczepem podniebienia W Kształcie Litery U i glossoptozą prowadzącą do obturacyjnego bezdechu i trudności z karmieniem. Sekwencja Pierre ‘ a Robina może być postrzegana jako część zespołów genetycznych (zespół delecji 22q11. 2, zespół Sticklera; opisany poniżej). (5)
istnieją setki zespołów genetycznych związanych z rozszczepami jamy ustnej, w tym nieprawidłowości cytogenetyczne (aneuploidy, mikrodelecje) i zaburzenia Jednogenowe (Mendelian). Potwierdzenie diagnozy genetycznej jest niezbędne do określenia rokowania i ustalenia ryzyka nawrotu choroby.
Aneuploidy, takie jak trisomia 13 i 18, mają silny związek z CL/CP. Trisomia 13 (inaczej zespół Pataua) jest związana z trzema kopiami chromosomu 13 lub niezrównoważonymi translokacjami Robertsona obejmującymi chromosom 13. Dzieci urodzone z tą chorobą zazwyczaj umierają w okresie noworodkowym. Cechy kliniczne obejmują rozszczep wargi i podniebienia, opóźnienie wzrostu, ciężkie wady wrodzone ośrodkowego układu nerwowego (w tym holoprosencefalia), małogłowie, mikroptalmia, coloboma tęczówki, brak oczu, zniekształcone uszy, polidaktylia, zaciśnięte pięści, wahliwe dolne stopy, wrodzone wady serca i wady układu moczowo-płciowego. Rozszczepy linii środkowej (w przeciwnym razie bardzo rzadko) można zaobserwować w trisomii 13 ze względu na ryzyko wad linii środkowej, w tym holoprosencefalii. Trisomia 18 (inaczej zespół Edwardsa) jest zwykle spowodowana trzema odrębnymi kopiami chromosomu 18 i wiąże się ze słabym wynikiem poporodowym. Cechy kliniczne obejmują rozszczep wargi i podniebienia, niepełnosprawność intelektualną, niepowodzenie w rozwoju, wrodzoną chorobę serca, hipertonię, mikrognatię, krótki mostek, nisko osadzone zniekształcone uszy, zaciśnięte dłonie, wahliwe dolne stopy i hipoplastyczne paznokcie. Trisomię 13 i 18 można łatwo potwierdzić lub wykluczyć wykonując analizę chromosomów (kariotypowanie).
zespoły mikrodelecji zazwyczaj obejmują delecję części chromosomu. Delecje te mogą być zbyt małe, aby można je było wykryć za pomocą standardowego kariotypowania i mogą wymagać wykrycia technologii Fish (fluorescence in situ hybridization) lub microarray. Dobrze znanym zespołem mikrodelecji związanym z rozszczepem podniebienia jest zespół delecji 22q11 .2 (znany również jako zespół Digeorge ‘ a/Velocardiofacial). Zaburzenia podniebienia, w tym niekompetencja welopharyngeal, rozszczepy podśluzówkowe, rozszczep błony naczyniowej i rozszczep podniebienia są widoczne u 69% osób z delecją 22q11.2 i mogą być częścią sekwencji Pierre Robin. Inne odkrycia kliniczne obejmują wrodzoną chorobę serca, utratę słuchu, cechy dysmorficzne, niedobór odporności, hipokalcemię, anomalie nerek, problemy z karmieniem, anomalie szkieletowe i zaburzenia psychiczne. Około 10% przypadków delecji 22q11.2 uważa się za rodzinne. Delecja segreguje się w sposób autosomalny dominujący.(6) zespół Wolfa-Hirschhorna, który jest spowodowany delecją w krótkim ramieniu chromosomu 4, jest również związany z rozszczepami jamy ustnej (u 25% do 50% osób dotkniętych chorobą). Charakterystyczne cechy twarzy (w tym wybitna glabella prowadząca do “wyglądu hełmu grecko-wojowniczego”), wrodzona choroba serca, niepełnosprawność intelektualna, napady padaczkowe, niepowodzenie w rozwoju, mikrognacja, tagi przeduszne lub doły i hipodontia mogą być również postrzegane jako część choroby.(7)
zaburzenia Jednogenowe z rozszczepami jamy ustnej obejmują między innymi zespół Sticklera, zespół Treachera Collinsa i zespół Van der Woude ‘ a. Zespół sticklera jest zaburzeniem kolagenowym z dziedziczeniem autosomalnym dominującym i, rzadziej autosomalnym recesywnym. Wspólne cechy obejmują rozszczep podniebienia (postrzegany jako część sekwencji Pierre ‘ a Robina lub bez mikrognatii), ubytek słuchu (odbiorczy i przewodzący), zmiany kostne (wczesne zapalenie stawów, dysplazja spondyloepiphyseal), anomalie oczne (wysoka krótkowzroczność, nieprawidłowości ciała szklistego) i charakterystyczne cechy twarzy (z niedorozwojem szczęki i mostka nosowego, retruzja śródstopia). Badania genetyczne na zespół Sticklera mogą być złożone, ponieważ mutacje w co najmniej sześciu genach zostały opisane u osób dotkniętych chorobą. Około 90% pacjentów z zespołem Sticklera ma mutacje w genie COL2A1 i ma autosomalną dominującą postać choroby.(8) Zespół Treachera Collinsa jest stanem autosomalnym dominującym charakteryzującym się rozszczepem podniebienia z rozszczepem wargi lub bez niego u 28% chorych. Inne nieprawidłowości obejmują hipoplazję kości jarzmowych i żuchwy, anomalie ucha zewnętrznego, coloboma dolnej powieki, przewodzeniowy ubytek słuchu, brak dolnych rzęs, przesunięcie włosów przedoczodołowych na policzki i zwężenie choanalu lub atrezję. Rozpoznanie zespołu Treachera Collinsa opiera się na wynikach badań klinicznych i radiologicznych. Opisano mutacje w co najmniej trzech genach, a mutacje w TCOF1 obserwowano u 78% do 93% pacjentów.(9) zespół Van der Woude ‘ a charakteryzuje się obecnością wrodzonych, Zwykle obustronnych, paramedycznych przetok dolnych warg (dołów), lub czasami małych kopców z układem zatokowym prowadzącym z gruczołu śluzowego wargi i rozszczepów jamy ustnej (w tym CL/CP i CP). Van der Woude jest stanem autosomalnym dominującym związanym z mutacjami w genie IRF6 (10). Badanie w Warunkach jedno-lub wielogenowych wymaga bezpośredniej analizy genu poprzez sekwencjonowanie i/lub analizę delecji / duplikacji (np. MLPA).
biorąc pod uwagę, że zespoły genetyczne z rozszczepem wargi i podniebienia mogą być związane z aneuploidies, mikrodelecjami/mikroduplikacjami chromosomów lub zaburzeniami jednogenowymi, testy genetyczne mogą być skomplikowanym procesem. Dokładna historia medyczna, rodowód trzech pokoleń, historia ciąży i badanie dysmorfologii przez genetyka klinicznego mogą wyjaśnić obraz kliniczny i umożliwić ukierunkowane badania genetyczne. Nowsze technologie, w tym mikromacierze, pozwolą na identyfikację małych mikrodelecji i mikroduplikacji, których wcześniej brakowało przy standardowym kariotypowaniu. Niestety, technika ta prowadzi również do identyfikacji delecji i duplikacji o nieznanym znaczeniu klinicznym, komplikując proces poradnictwa genetycznego. Badanie w kierunku zaburzeń jednogenowych lub zaburzeń Mendlowskich wymaga klinicznej dostępności badań genetycznych dla pożądanego genu. Może to być również kosztowne, jeśli nie jest objęte ubezpieczeniem medycznym. Nowe technologie, takie jak sekwencjonowanie następnej generacji, sekwencjonowanie exome, lub sekwencjonowanie genomu (znany zbiorczo jako testy genomowe) stały się klinicznie dostępne. Analizując jednocześnie setki do tysięcy genów, testy te znacznie zwiększają moc diagnostyczną i wydajność. W porównaniu z innymi technikami, testy te mogą dostarczyć odpowiedzi szybciej i w bardziej opłacalny sposób. W dziedzinie badań sekwencjonowanie egzomu i genomu doprowadziło do identyfikacji nowych genów, a także rozszerzenia cech klinicznych i spektrum mutacji genetycznych. Podobnie jak w przypadku technologii mikromacierzy, testy genomowe mogą wykryć zespoły, które nie są związane z prezentacją pacjenta i / lub powodem badania. Biorąc pod uwagę nieodłączne zawiłości badań genetycznych, konieczna jest świadoma zgoda.
wniosek
chociaż rozszczep wargi i podniebienia jest izolowaną anomalią w większości przypadków, istnieje silny związek między rozszczepami jamy ustnej a innymi anomaliami i zespołami genetycznymi. Ocena genetyczna przez genetyka klinicznego i doradcę genetycznego jest niezbędna do przewidywania wytycznych i określenia ryzyka nawrotu. Badania genetyczne, które wymagają świadomej zgody, mogą być koordynowane i interpretowane podczas oceny genetycznej.
Anya Revah, MS, jest starszym doradcą genetycznym w oddziale Genetyki Medycznej w Maimonides Infants and Children ‘ s Hospital w Brooklynie w Nowym Jorku. Jest również aktywnym członkiem multidyscyplinarnego zespołu Maimonides Medical Center i Kings County Hospital Cleft Lip and Palate. Ukończyła studia magisterskie z zakresu poradnictwa genetycznego na Uniwersytecie Bostońskim w Bostonie w stanie Massachusetts.
1. Sadler TW. Embriologia medyczna langmana. Dziewiąta Edycja. Strony 390-395.
2. Parker SE, Mai CT, Canfield MA, Rickard R, Wang Y, Meyer RE, Anderson P, Mason CA, Collins JS, Kirby RS, Correa A. dla Krajowej Sieci zapobiegania wadom wrodzonym. Zaktualizowane krajowe szacunki częstości występowania urodzeń dla wybranych wad wrodzonych w Stanach Zjednoczonych. 2004-2006. Badania nad wadami wrodzonymi (część A): Teratologia kliniczna i molekularna 2010; 88: 1008-1016.
3. Fraser FC. Genetyka rozszczepu wargi i rozszczepu podniebienia. Am. J. Hum. Genet. 1970;22: 336–352.
4. Van Rooij IA, OCKE MC, et al. Perikonceptualne spożycie kwasu foliowego przez suplement i spożycie pokarmu zmniejsza ryzyko wystąpienia rozszczepu wargi bez zespołu z rozszczepem podniebienia lub bez niego. Prev Med 2004; 39: 689-694.
5. Tan TY. Kilpatrick N, Farlie PG. Rozwojowe i genetyczne perspektywy sekwencji Pierre ‘ a Robina. Am. J. Med. Genet. 2013; 163C: 295-305.
6. McDonald-McGinn DM, Emanuel BS, Zackai EH. Zespół delecji 22q11.2. Wrzesień 23, 1999. . W: Pagon RA, Adam MP, Ardinger HH, et al., redakcja. Generaeviews . 1993-2014 Dostępny od: http://www.ncbi.nlm.nih.gov/books/NBK1523/.
7. Battaglia a, Carey JC, South ST, et al. Zespół Wolfa-Hirschhorna. Kwiecień 29, 2002. . W: Pagon RA, Adam MP, Ardinger HH, et al., redakcja. Generaeviews . 1993-2014 Dostępny od: http://www.ncbi.nlm.nih.gov/books/NBK1183/
8. Robin NH, Moran RT, Ala-Kokko L. Stickler Syndrome. Jun. 9, 2000. . W: Pagon RA, Adam MP, Ardinger HH, et al., redakcja. Generaeviews . 1993-2014 Dostępny od: http://www.ncbi.nlm.nih.gov/books/NBK1302/
9. Katsanis SH, Jabs EW. Zespół Treachera Collinsa. Lip. 20, 2004. . W: Pagon RA, Adam MP, Ardinger HH, et al., redakcja. Generaeviews . Seattle (WA): University of Washington, Seattle; 1993-2014. Dostępny od: http://www.ncbi.nlm.nih.gov/books/NBK1532/.
10. Schutte BC, Saal HM, Goudy S, et al. Zaburzenia związane z IRF6. Październik 30, 2003. . W: Pagon RA, Adam MP, Ardinger HH, et al., redakcja. Generaeviews . 1993-2014 Dostępny od: http://www.ncbi.nlm.nih.gov/books/NBK1407/