mukopolisacharydozy: wczesne objawy diagnostyczne u niemowląt i dzieci
mukopolisacharydozy (MPS) to grupa klinicznie heterogenicznych chorób spowodowanych niedoborami enzymów lizosomalnych wymaganych do rozpadu glikozaminoglikanów (GAGs).
MPS charakteryzują się postępującymi i układowymi objawami klinicznymi. Pomimo ich biochemicznej i genetycznej heterogeniczności, różne typy mają kluczowe cechy kliniczne w różnych kombinacjach, w tym dysplazję stawów i szkieletów ze sztywnością (z wyjątkiem MPS IV, gdzie występuje wiotkość) i ból, grube rysy twarzy, zmętnienie rogówki, przepukliny pachwinowe lub brzuszne, nawracające infekcje górnych dróg oddechowych, choroby zastawek serca, zespół cieśni nadgarstka i zmienne zaangażowanie neurologiczne. Cechy te pojawiają się zwykle w pierwszych miesiącach życia dla form ciężkich i we wczesnym dzieciństwie dla form bardziej atenuowanych, ale często są niedoceniane i ogólnie brane pod uwagę tylko wtedy, gdy są wyraźnie widoczne.
rzadkość tych zaburzeń i zmienność prezentacji klinicznej często prowadzi do opóźnienia diagnostycznego; może to wynosić od miesięcy, dla ciężkich postaci, do lat, dla atenuowanych (patrz Rigoldi i wsp. w tym suplemencie). Niemniej jednak, ze względu na szybki postęp i pilną potrzebę interwencji w ciężkich prezentacjach, nawet miesiące opóźnienia w diagnozie mogą przynieść katastrofalne wyniki dla przyszłego zdrowia pacjenta.
naszym celem jest podkreślenie w tym przeglądzie bardzo wczesnych objawów ciężkich postaci, które powinny ostrzec pediatrę o ich pierwszym pojawieniu się.
jakich objawów przedmiotowych/podmiotowych możemy się spodziewać u dzieci z ciężkimi postaciami MPS?
objawy podmiotowe i przedmiotowe sugerujące różne formy MPS przedstawiono w tabeli 1 w zależności od wieku. W pierwszych 6 miesiącach życia u pacjentów z MPS można zaobserwować przepukliny pachwinowe, nienormalnie częste infekcje dróg oddechowych, zapalenie ucha i organomegalię, szczególnie w MPS I (zespół Hurlera), który ma najbardziej przedwczesny ciężki początek. Ponadto od 6 do 12 miesięcy niemowlęta te bardzo często rozwijają gibbusa (kifozę torako-lędźwiową), szmer serca, przepuklinę pępkową, łagodną hipotonię i opóźnienie wzrostu . Mogą one również mieć typowy szorstki wygląd twarzy (rys. 1). MPS II, VI i VII mogą występować z podobnym wczesnym fenotypem. Pacjenci z MPS IV mają wczesny fenotyp szkieletu z pojawieniem się dysplazji stawów biodrowych w pierwszych miesiącach życia i gołębia (pectus carinatum) w pierwszych 12-18 miesiącach. Niemowlęta MPS II występują z podobnym zaangażowaniem, ale z późniejszym początkiem. W drugim roku życia dzieci z zespołem MPS i Hurler rozwijają opóźnienie poznawcze, podczas gdy pacjent MPS II może być zidentyfikowany tylko z powodu przerostu, częstych infekcji dróg oddechowych i wielu operacji ; typowe rysy twarzy pojawiają się dopiero później. U niektórych pacjentów z MPS II rozwija się również nadpobudliwość między 18 a 24 miesiącem. W trzecim roku życia nadpobudliwość i opóźnienie poznawcze są łatwo rozpoznawalne w MPS II i MPS III.
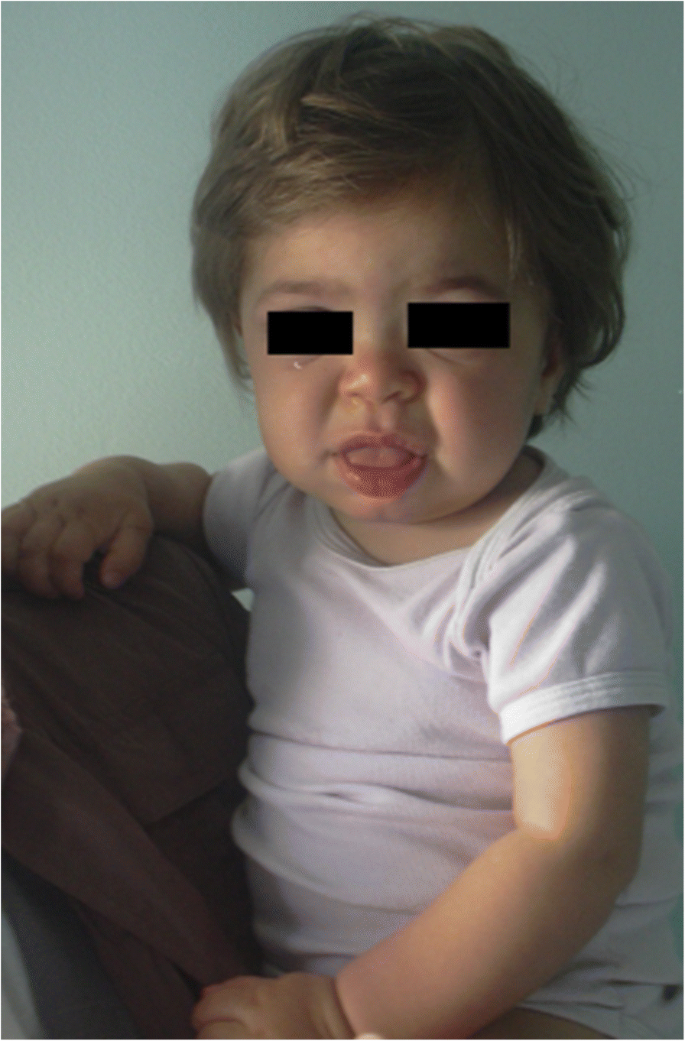
MPS IH u 1-letniego pacjenta. Grube twarze: płaski most nosowy, makroglosja, przedni mostek
podsumowując, objawy kostne są typowe, a czasami przedwczesne. Grube twarze i opóźnienie poznawcze nie są wczesnymi widocznymi oznakami.
jakie są różne prezentacje różnych typów posłów?
MPS z zaangażowaniem somatycznym i poznawczym
Mukopolisacharydoza typu I (MPS i; OMIM #252800) (Fig. 1, 2, 3, 4 i 5) jest spowodowany niedoborem lizosomalnej hydrolazy α-l-iduronidazy, co prowadzi do wielosystemowej akumulacji dwóch Gag: siarczanu dermatanu (DS) i siarczanu heparanu (HS) . Ze względu na stopień zaawansowania, MPS i tradycyjnie dzieli się na trzy fenotypy kliniczne, jednak formy te stanowią kontinuum stopnia zaawansowania choroby. Zespół Hurlera, najcięższa postać, zwykle prezentuje się w pierwszym roku życia. Dotknięte dzieci szybko rozwijają znaczne upośledzenie funkcji poznawczych i choroby somatyczne w wielu układach narządów, co prowadzi do śmierci w ciągu pierwszej dekady bez leczenia. Atenuowane formy MPS I, znane odpowiednio jako zespół Hurler-Scheie i zespół Scheie, charakteryzują się późniejszym wystąpieniem objawów, dłuższą oczekiwaną długością życia i łagodnym lub bez udziału ośrodkowego układu nerwowego (OUN). Dziedziczenie jest autosomalne recesywne, a w co najmniej 50% przypadków przewidywanie fenotypu jest możliwe na podstawie genotypu, podczas gdy w pozostałych 50% wykrywane są nowe mutacje . Częstość występowania wynosi około 1 na 100 000 żywych urodzeń .

MPS IH. Gibbus u 9-miesięcznego pacjenta. b RTG kręgosłupa u 3-letniego pacjenta. C T2-ważony obraz rezonansu magnetycznego kręgosłupa wykazujący znaczącą kifozę i zwężenie kanału kręgowego

MPS IH. Rentgenowskie dowody dysostozy multiplex. pogrubienie żeber U 3-letniego pacjenta i dysplazja stawu biodrowego b u innego pacjenta w wieku 18 miesięcy

MPS IH: pazur dłoni u 18-miesięcznego pacjenta; RTG dłoni B.

MPS IH. zgrubienie kości korowej czaszki i nieprawidłowa Budowa” J-shaped ” sella turcica. Poszerzenie przestrzeni okołoporodowej w obrazach rezonansu magnetycznego B I c T-2 u pacjenta w wieku 17 miesięcy
Mukopolisacharydoza typu II (MPS II; OMIM #309900) (rys. 6), znany również jako zespół Huntera, jest wynikiem niedoboru enzymu iduronian-2-sulfatazy (I2S) z konsekwencją nagromadzenia GAG DS i HS . Częstość występowania wynosi około 1 na 140 000–156 000 żywych urodzeń samców . Warto zauważyć, że jest to jedyny MPS związany z chromosomem X, A żeńskie nosiciele są zwykle bezobjawowe; jednak mogą być wyjątkowo dotknięte z powodu nieprawidłowości chromosomu X, homozygotyczności lub zniekształconej inaktywacji X. Ekspresja fenotypowa obejmuje również szerokie spektrum nasilenia klinicznego. W ciężkich przypadkach współistnieją wyraźne postępujące zaangażowanie neurologiczne i choroby somatyczne, a śmierć następuje zwykle w drugiej dekadzie życia. Inni pacjenci z minimalnymi lub żadnymi dysfunkcjami neurologicznymi mogą stanowić poważne zaangażowanie somatyczne, podczas gdy na przeciwległym końcu spektrum są pacjenci z minimalnymi objawami somatycznymi i normalną inteligencją, którzy przetrwają do dorosłości . Wczesne objawy są wspólne dla ciężkich postaci MPS I I II.

MPS II. 3-letni pacjent z tylko łagodnymi charakterystycznymi rysami twarzy
sporadyczne przypadki hydrops fetalis są opisane w MPS I, chociaż ogólnie dzieci te wydają się normalne po urodzeniu. Pierwsze oznaki MPS I pojawiają się zwykle w pierwszych miesiącach życia, natomiast te w MPS II pojawiają się nieco później . Kiely et al. , w retrospektywnym badaniu z udziałem 55 pacjentów Hurler, zaobserwowano, że objawy ze strony układu oddechowego, trudności z karmieniem, przepuklina pachwinowa i zapalenie ucha środkowego pojawiły się w średnim wieku ≤ 6 miesięcy, a kifoza, zaburzenia serca, powiększony obwód głowy, hipotonia, organomegalia, zmętnienie rogówki, ograniczenie stawów i przepuklina pępkowa pojawiły się między 6 A 12 miesiącem. Przepuklina pępkowa jest jedną z najwcześniej występujących cech w MPS I I II, a przepukliny pachwinowe występują u około 60% chorych .
Inną częstą początkową wskazówką dla MPS I I II mogą być nawracające infekcje górnych dróg oddechowych ze zwiększoną wydzieliną. Częste zapalenie ucha, przewlekły nawracający nieżyt nosa i uporczywe wydzieliny z nosa bez oczywistego zakażenia nie są specyficzne, ale mogą pomóc w wzmocnieniu podejrzenia diagnostycznego, jeśli związane z bardziej specyficznymi objawami. Przechowywanie GAG w obrębie gardła oro prowadzi do powiększenia migdałków i migdałków migdałkowych i przyczynia się do powikłań górnych dróg oddechowych. Innym częstym stwierdzeniem jest głośne oddychanie, szczególnie w nocy, związane w późniejszych stadiach z obturacyjnym bezdechem sennym. W wyniku częstych infekcji ucha środkowego i dysostozy kosteczek ucha środkowego, utrata słuchu jest częsta, zarówno z deficytami przewodzącymi, jak i odbiorczymi . Tracheobronchomalacja jest powszechnie obserwowana i może prowadzić do ostrej niedrożności dróg oddechowych lub zapaści, przy czym jest to jedna z głównych przyczyn śmierci w kolejnych latach .
Adenoidektomia, tonsillektomia i tympanostomia wraz z naprawą przepukliny są częstszymi i wczesnymi zabiegami chirurgicznymi u pacjentów z MPS I I II, często wykonywanymi przed rozpoznaniem (w ponad 50% przypadków).
nawracająca biegunka jest dość powszechna we wczesnych stadiach MPS I, może być również obecna u pacjentów z MPS II z neurologicznym zaangażowaniem, podczas gdy jest rzadka u pacjentów o łagodnym przebiegu choroby .
deformacja Gibbusa (kifoza grzbietowo-lędźwiowa) (rys. 2) ujawnia się klinicznie u mediany wieku 8 lat.Od 6 miesięcy do 1 roku w MPS I, co stanowi dość specyficzny i przedwczesny wskaźnik MPS w ogóle. Ciała kręgowe wydają się spłaszczone i dzioby, co może prowadzić do późniejszych powikłań, takich jak uwięzienie nerwu rdzeniowego, ostre uszkodzenie kręgosłupa i niestabilność potyliczna; dlatego należy zachować szczególną ostrożność, aby zapobiec zwichnięciu stawu atlantoosialnego podczas znieczulenia ogólnego i zabiegu chirurgicznego. Po 1 roku życia, przykurcze stawów i genu valgum stanowią inne częste objawy upośledzenia stawów i postępującej dysplazji szkieletowej, które są łatwo wykrywalne w wieku powyżej 2 lat u wszystkich niemowląt dotkniętych MPS. Wszystkie te objawy kostne razem, typowe dla MPS, nazywane są dysostosis multiplex. Pogrubienie żeber można zobaczyć na zdjęciach radiologicznych (rys. 3a), kości długie są krótkie z szerokimi wałkami, a kolana są podatne na koślawość i zniekształcenia warg. Dysostoza paliczkowa i zgrubienie błony maziowej prowadzą do charakterystycznej deformacji dłoni pazura (rys. 4), co jest kolejną typową cechą, która może wzbudzić podejrzenie choroby. Dysplazja stawu biodrowego jest często powodem, dla którego dzieci te mogą być po raz pierwszy obserwowane przez specjalistów ortopedycznych. Zazwyczaj miednica jest słabo uformowana, głowy kości udowej są małe, a coxa valga jest powszechna, z postępującą i wyniszczającą deformacją biodra (rys. 3b).
istnieje znacząca różnica między MPS I i MPS II; w MPS I wzrost szkieletu zwalnia o 3 lata, podczas gdy pacjenci MPS II wykazują przerost w pierwszym 5-6 roku życia , spowalniając ich wzrost w późniejszym okresie .
zaostrzenie rysów twarzy (szeroki nos z rozszerzonymi nozdrzami, wydatne grzbiety nadoczodołowe, duże zaokrąglone policzki, grube wargi, powiększony wystający język) spowodowane magazynowaniem gagów w tkankach miękkich okolicy orofacial i kości twarzy staje się widoczne w ciągu pierwszych 2 lat, w średnim wieku 1,1 roku dla choroby Hurlera i od 18 miesięcy do 4 lat w ciężkiej postaci choroby Huntera , chociaż subtelne zmiany fenotypowe można wykryć wcześniej (już w drugiej połowie pierwszego roku) przez pediatrów ze szczególnym doświadczeniem (rys. 1). Macrocephaly staje się coraz bardziej widoczne, a scafocefalia jest powszechne (rys. 5), ale małogłowie jest również możliwe u pacjentów Hurler . Hirsutyzm twarzy i ciała jest zawsze widoczny w wieku 24 miesięcy . Skóra może być pogrubiona i nieelastyczna. Warto zauważyć, że u niektórych pacjentów z MPS II rozwija się charakterystyczne uszkodzenie skóry, które jest opisane jako białe grudki (kamyczkowate) na górnej części pleców i bokach ramion, patognomoniczne zespołu Huntera .
kardiomiopatia i postępujące pogrubienie i usztywnienie listków zastawkowych u pacjentów z MPS I mogą prowadzić do mitralnej i, rzadziej, niedomykalności aorty . Szmer może być wykryty w pierwszych miesiącach życia i czasami jest zgłaszany jako pierwszy objaw choroby . Zaangażowanie serca występuje również u prawie wszystkich pacjentów z MPS II, ale zaczyna się to w wieku około 5 lat . Choroba zastawki może prowadzić do przerostu prawej i lewej komory i niewydolności serca.
wypukłość brzucha spowodowana postępującą hepatosplenomegalią jest łatwo widoczna po 6 miesiącu życia, chociaż przechowywanie gagów w wątrobie i śledzionie nie prowadzi do dysfunkcji narządów .
zmętnienie rogówki występuje u prawie wszystkich osób z MPS I przed 15 miesiącem życia , potencjalnie powodując poważne zaburzenia widzenia. Przeciwnie, zmętnienie rogówki nie jest typową cechą zespołu Huntera, ale badanie lampą szczelinową może ujawnić dyskretne zmiany rogówki, które nie wpływają na widzenie. Dysfunkcję siatkówki można wykryć, natomiast jaskra nie jest częstym stwierdzeniem.
wczesny rozwój psychomotoryczny jest zwykle wykrywany jako normalny, ale w MPS i opóźnienie rozwoju jest zwykle oczywiste w wieku 18 miesięcy, z postępującym spadkiem psychicznym prowadzącym do poważnej niepełnosprawności intelektualnej. U tych dzieci umiejętności językowe są bardzo ograniczone. Globalne opóźnienie etapów rozwojowych w MPS II staje się widoczne już po dwóch latach . Jednak wiodącą cechą neurologiczną ciężkiej choroby Huntera są wyraźne zaburzenia zachowania, takie jak nadpobudliwość, upór i agresywność, które zazwyczaj nie są obserwowane u osób z atenuowanym fenotypem .
Mukopolisacharydoza typu VII (MPS VII; OMIM #253220), znany również jako zespół Sly, jest ultra-rzadkim MPS charakteryzującym się niedoborem aktywności β-glukuronidazy, z lizosomalnym magazynowaniem siarczanu chondroityny (CS), DS i HS, prowadzącym do dysfunkcji komórkowych i narządów. Pierwszy pacjent został opisany przez Sly et al. w 1973 roku w piśmiennictwie opisano dotychczas 145 pacjentów.
prezentacja kliniczna i progresja choroby MPS VII obejmuje szerokie spektrum nasilenia, od wczesnych, ciężkich, wielosystemowych objawów i śmierci w pierwszych miesiącach, do łagodniejszego fenotypu z późniejszym początkiem, prawidłową lub prawie normalną inteligencją i dłuższym przeżyciem .
cechy fenotypu pacjentów z MPS VII przypominają cechy MPS I i MPS II (niski wzrost, dysplazja szkieletowa, makrocefalia, przerost dziąseł, nawracające infekcje ucha, hepatosplenomegalia, przepukliny, zajęcie serca, upośledzenie czynności płuc i zaburzenia funkcji poznawczych). Nieoczekiwanie wysoki odsetek opisanych pacjentów (41%) miał w wywiadzie nieimmunologiczny noworodek hydrops fetalis (NIHF) . Pomimo prenatalnego początku choroby u tych pacjentów, 13 z 23 przeżyło niemowlęctwo z łagodnym do pośredniego przebiegiem do późnej nastolatków. Tak więc obecność NIHF sama w sobie nie przewiduje możliwego nasilenia przebiegu klinicznego, jeśli pacjent przeżyje niemowlęctwo. Innym wczesnym objawem jest makrokrania, natomiast zajęcie zastawek serca i powtarzające się infekcje górnych i dolnych dróg oddechowych pojawiają się w pierwszych 2 latach życia. Umiarkowane upośledzenie umysłowe i postępująca utrata słuchu z zaburzeniami mowy są również widoczne z czasem.
podsumowując, ciężkie formy MPS mam bardzo wczesny początek wielosystemowy (w ciągu 6 miesięcy życia); MPS II jest podobny, ale z późniejszym początkiem, a MPS VII ma początek prenatalny z częstym NIHF i wczesnym ciężkim zajęciem górnych i dolnych dróg oddechowych.
MPS z głównie zaangażowaniem neurologicznym i poznawczym
mukopolisacharydozy typu III (MPS III, znany również jako zespół Sanfilippo; Fig. 7) ma dominującą prezentację neurologiczną. MPS III jest zaburzeniem magazynowania lizosomalnego spowodowanym niedoborem jednego z czterech enzymów biorących udział w katabolizmie HS. Znane są cztery różne podtypy—MPS III typ A (OMIM #252900), Typ B (OMIM #252920), Typ C (OMIM #252930) i typ D (OMIM #252940)—każdy z powodu innego niedoboru enzymu: Typ A, Gen sulfamidazy/SGSH; Typ B, Gen alfa-N-acetyloglukozaminidazy/NAGLU; Typ C, N-acetylotransferaza Alfa-glukozaminidu/HGSNAT Gen; typ D, N-acetilglucosamina-6-solfato sulfatasi/Gen GNS). Wszystkie cztery typy (od A do D) mają dziedziczenie autosomalne recesywne. MPS III jest najczęstszym z MPS o szacowanej częstości występowania między 0,3 A 4.1 przypadki na każde 100 000 noworodków, w zależności od podtypu i rasy .

MPS IIIA. Ten sam pacjent w wieku a 2,5 i B 5 lat. Zwróć uwagę na inny wyraz twarzy, pokazujący postęp zaburzeń neurologicznych
pacjenci z zespołem Sanfilippo wykazują postępujące zaburzenia poznawcze w drugim i trzecim roku życia. W przeciwieństwie do większości MPS, cechy somatyczne są stosunkowo mniej widoczne, a zaangażowanie OUN jest widoczne. Opóźnienie rozwoju mowy jest zwykle pierwszym objawem zauważonym przez rodziców, ale głównymi problemami mogą być problemy behawioralne (nadpobudliwość, niespokojne i agresywne zachowanie, zaburzenia snu), a następnie postępujący spadek psychiczny . Objawy te mogą powodować błędne rozpoznanie, takie jak zaburzenia zachowania, zespół nadpobudliwości psychoruchowej z deficytem uwagi (ADHD) i zaburzenia ze spektrum autyzmu . Może również występować padaczka.
zaburzenia snu, których częstość występowania jest zgłaszana do 80-90% , są wspólną cechą. Składają się one z trudności w zasypianiu i częstego nocnego przebudzenia, z całkowitym odwróceniem rytmu dzień-noc u niektórych pacjentów.
wszystkie te przejawy są bardzo trudne do opanowania i mają ogromny wpływ psychologiczny na jakość życia całej rodziny.
ogólnie, chociaż fenotyp może być bardzo podobny w czterech podtypach, przebieg kliniczny w Sanfilippo B I C wydaje się charakteryzować mniej ciężkim fenotypem .
objawy Nieneurologiczne są zwykle mniej wyraźne w MPS III niż w innych MPS. Nawracające infekcje ucha, nosa i gardła obserwuje się w młodszym wieku. Nawracające zapalenie ucha środkowego, wady kosteczek ucha środkowego i nieprawidłowości w uchu wewnętrznym może powodować głuchotę. Nadmiar gęstej wydzieliny i zmiany anatomiczne mogą powodować niedrożność dróg oddechowych. W pierwszych latach życia często występuje biegunka, natomiast u starszych pacjentów częściej występują zaparcia.
badanie fizykalne może wykazać grube rysy twarzy, chociaż są one mniej wyraźne. Pacjenci mają makrocefalię, szerokie brwi z rozchyleniem przyśrodkowym i synophrys, włosy są zwykle suche i szorstkie, a hipertrichoza jest powszechna. U młodszych pacjentów często obserwuje się łagodne powiększenie wątroby oraz przepuklinę pępkową i pachwinową. Rzadkie i często łagodne przykurcze występują głównie w łokciach. Wzrost jest zaburzony u około połowy pacjentów w wieku powyżej 12 lat .
podsumowując, choroba MPS III lub Sanfilippo ma wyraźny udział neurologiczny z bardzo łagodnymi objawami somatycznymi. Małe dzieci w wieku 2-3 lat rozwijające nadpobudliwość, ADHD i agresywne zachowanie mogą być podejrzane o MPS III.
MPS z przeważającym zaangażowaniem somatycznym
MPS IV i MPS VI występują tylko z zaangażowaniem somatycznym. Są to Stany postępujące, które oszczędzają zdolności intelektualne i wpływają głównie na szkielet (MPS IVA)lub wszystkie narządy / układy ciała (MPS VI). Dziedziczenie jest autosomalne recesywne. Tempo nasilania się objawów różni się u osób dotkniętych chorobą.
Mukopolisacharydoza IV (MPS IV, znany również jako zespół Morquio; rys. 8) przedstawia się jako dwa odmienne genetycznie zaburzenia, z których każdy ma inny niedobór enzymu: MPS Iva (zespół Morquio a; OMIM #253000) z powodu niedoboru N-acetylogalaktozaminy-6-sulfatazy (Gen GALNS), powodujący akumulację siarczanu keratanu (KS) i CS ; i MPS IVB (zespół Morquio B; OMIM #253010) z powodu niedoboru aktywności beta-galaktozydazy prowadzącej do wydalania KS i oligosacharydów w moczu, jak u pacjentów z GM1. MPS IVB jest rzeczywiście alleliczny do różnych form gangliozydozy GM1, ale brakuje pogorszenia psychomotorycznego obserwowanego w gangliozydozie GM1 .

MPS IVA. 4-letni pacjent z przednio-tylnym i bocznym prześwietleniem b, wykazującym żebra w kształcie łopatki oraz spłaszczone i zaokrąglone kręgi z typowym” przednim dziobem”
u pacjentów z ciężkim MPS IVA bez wykrywalnej aktywności enzymatycznej pierwsze objawy pojawiają się zwykle w pierwszym roku życia, chociaż rzadko są one diagnozowane przed 4-5 rokiem życia. Informacje na temat historii naturalnej pacjentów z Morquio A pochodzą głównie z dwóch obszernych zbiorów danych . Zmniejszenie tempa wzrostu rozpoczyna się w wieku około 18 miesięcy, a wzrost zatrzymuje się w wieku około 7 lub 8 lat. W przeciwieństwie do innych MPS, stawy Morquio a są zwykle luźne i bardzo elastyczne (hipermobilne), ale niektóre stawy (Zwykle biodra i ramiona) mogą mieć ograniczony zakres ruchu. Pacjenci z takim zajęciem szkieletu mogą otrzymać diagnozę lub poddać się ocenie dysplazji spondyloepiphyseal, pseudoachondroplazji, wielokrotnej dysplazji nasadowej lub obustronnej chorobie Legg-Calvé-Perthesa .
cechą stałą jest hipoplazja odontoidów; w ten sposób może dojść do zwichnięcia stawu potyliczno-potylicznego, powodując ucisk i uszkodzenie opuszki i rdzenia kręgowego, powodując paraliż, a nawet śmierć, jeśli Stabilizacja szyjki macicy nie zostanie wykonana wcześnie.
pacjenci MPS IVB mają bardzo podobny objaw kliniczny, ale z mniej widocznym niskim wzrostem. Mają lekko grube rysy twarzy, utratę słuchu, zmętnienie rogówki, zastawkowe choroby serca i częste infekcje górnych dróg oddechowych. Mogą również występować z przepukliną pachwinową i łagodną hepatomegalią. Cechy dysplastyczne szkieletu obejmują platyspondylię, hipoplazję odontoidalną z możliwością podwichnięcia szyjki macicy, skoliozę kipho, coxa valga i zwężone skrzydła biodrowe .
Mukopolisacharydoza VI (MPS VI, znany również jako zespół Maroteaux-Lamy; OMIM #253200; Fig. 9) jest spowodowany mutacjami patogennymi w genie arylosulfatazy B (ARSB) zlokalizowanym na chromosomie 5. W konsekwencji zmniejszona lub nieobecna aktywność enzymu arylosulfatazy B (asb) (4-sulfatazy N-acetylogalaktozaminy) upośledza degradację DS determinując jego akumulację komórkową .

MPS VI. 2-letni chłopiec z wyraźną szorstką twarzą i dysostozą szkieletową
pacjenci bez wykrywalnej aktywności enzymów mają ciężką postać MPS VI i wykazują objawy we wczesnym dzieciństwie. Ich fenotyp somatyczny jest podobny do zespołu Hurlera. W większości przypadków, w wieku 2 lub 3 lat, ciężkie i postępujące uszkodzenie szkieletu, o nazwie dysostosis multiplex, staje się oczywiste. Ta dysplazja szkieletowa obejmuje dysplazję stawu biodrowego z dysplastyczną głową kości udowej, nieprawidłowe kręgi, nieregularne obojczyki, niedorozwój dystalnej kości łokciowej i promieniowej oraz dysplastyczne i krótkie kości śródręcza; kości nadgarstka i stępu są hipoplastyczne i mają nieregularny profil. Prędkość wzrostu często zwalnia po pierwszym roku życia, a ostateczny wzrost jest zazwyczaj niższy niż 120 cm. Podobnie jak w innych MPS, wczesnym objawem są grube rysy twarzy. Ponadto pacjenci mają typowy wystający brzuch, powiększenie wątroby, przepuklinę pępkową i / lub pachwinową oraz zajęcie serca we wczesnym dzieciństwie. Chociaż nie występuje pierwotna niepełnosprawność intelektualna, wodogłowie można zaobserwować u niektórych pacjentów z wtórnym nadciśnieniem śródczaszkowym i brodawczakiem. Pectus carinatum, sztywne i skurczone stawy, skolioza i kifoza oraz kompresja szyjnego rdzenia kręgowego pogarszają się z czasem .
podsumowując, posłowie IV i VI nie wykazują zaangażowania OUN. MPS IV jest unikalnym MPS w wykazywaniu wiotkości stawów, podczas gdy ciężki początek MPS VI jest podobny do obserwowanego w MPS I.