A genética da fenda labial e palato
 Intro / abstractCleft lip com ou sem fenda palato é uma anomalia congénita complexa que pode ser isolada ou vista juntamente com outras malformações. Pode também fazer parte do fenótipo de uma síndrome genética. Este artigo serve como uma revisão da prevalência de fissura labial e palato, riscos de recorrência e riscos para outras anomalias congênitas. Serão exploradas síndromes genéticas e exposições teratogénicas que se sabe estarem associadas a fissuras orais. Além disso, serão discutidos os testes genéticos comumente solicitados no ambiente pediátrico de genética clínica para a avaliação do paciente com fenda labial e palato.
Intro / abstractCleft lip com ou sem fenda palato é uma anomalia congénita complexa que pode ser isolada ou vista juntamente com outras malformações. Pode também fazer parte do fenótipo de uma síndrome genética. Este artigo serve como uma revisão da prevalência de fissura labial e palato, riscos de recorrência e riscos para outras anomalias congênitas. Serão exploradas síndromes genéticas e exposições teratogénicas que se sabe estarem associadas a fissuras orais. Além disso, serão discutidos os testes genéticos comumente solicitados no ambiente pediátrico de genética clínica para a avaliação do paciente com fenda labial e palato.
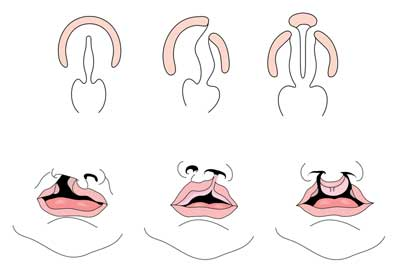
fenda labial com ou sem fenda palato (CL/CP) difere de uma fenda palato isolada (CP) em níveis embrionários, epidemiológicos e genéticos. O lábio leporino tipicamente resulta da proeminência maxilar e proeminência nasal medial não se fundindo entre a quinta e sexta semana de desenvolvimento embrionário. O desenvolvimento normal do palato resulta da formação do palato primário e do palato secundário. O palato primário é formado nas semanas seis a sete pelo desenvolvimento e fusão dos processos nasal medial, nasal lateral e maxilar. O palato secundário se origina das prateleiras palatais (que se desenvolvem a partir dos processos maxilares emparelhados do primeiro arco branchial) tornando-se horizontal e fundindo, formando os palatos duros e macios em torno da nona semana de desenvolvimento embrionário. As prateleiras também se fundem com o palato primário e septo nasal. (1)
as fissuras orais são um dos defeitos de nascença mais comuns observados no berçário neonatal, com uma prevalência global de 1.6 por mil recém-nascidos em todo o mundo, com CL/CP visto em aproximadamente um por mil nascimentos e CP visto em 0,6 por mil nascimentos. (2) Há uma maior frequência de CL/CP em indivíduos de ascendência asiática, africana e Nativa Americana. CL / CP também é mais comum em homens. Em contraste, não há diferença significativa na incidência de CP entre diferentes origens étnicas, e CP é mais comum em mulheres. (3) os riscos de recorrência dentro de uma família dependem do facto de a fenda ser isolada (sem outros resultados clínicos presentes) ou vista como parte de uma síndrome genética. A maioria dos casos de fissuras orais são isolados (aproximadamente 80%). Acredita-se que fissuras isoladas tenham herança multifatorial: são devido a uma combinação de múltiplos fatores, tanto genéticos como ambientais. O risco de recorrência (Tabela 1) aumenta quando há mais de um parente afetado. O risco de recorrência também aumenta o mais grave o defeito é.

Cleft lip and palate pode ser visto com outras anomalias congénitas. A probabilidade de uma etiologia genética ou teratogênica aumenta as anomalias congênitas com as quais um paciente apresenta. A presença de outras questões, tais como deficiência intelectual, problemas comportamentais, tais como autismo, características dismórficas, ou outras preocupações médicas também fará uma desordem genética ou uma exposição teratogênica mais provável. Aproximadamente 13% dos indivíduos com fissura labial terão outras preocupações médicas ou anomalias. O número aumenta para 37% com fenda labial e palato e para 47% apenas com fenda palato.
exposição pré-natal a agentes teratogénicos (tais como a talidomida, anticonvulsivantes, álcool, ácido retinóico e cigarros) e doenças maternas (tais como diabetes, rubéola e deficiência em folato) têm demonstrado aumentar o risco de fissuras orais. A presença de bandas amnióticas também aumenta o risco de fissuras. Sabe-se que a suplementação com ácido fólico Periconceptual reduz o risco de fissuras orais.A sequência Pierre Robin é uma anomalia craniofacial caracterizada por hipoplasia mandibular ou micrognatia, palato de fenda secundário em forma de U, e glossoptose levando a apneia obstrutiva e dificuldades de alimentação. A sequência de Pierre Robin pode ser vista como parte das síndromes genéticas (síndrome de deleção 22q11.2, síndrome de Stickler; descrito abaixo). (5)
existem centenas de síndromes genéticas associadas a fissuras orais, incluindo anomalias citogenéticas (aneuploidias, microdeletções) e distúrbios de gene único (Mendeliano). Confirmar um diagnóstico genético é essencial para determinar o prognóstico e estabelecer um risco de recorrência.
Aneuploidies such as Trisomy 13 and 18 have a strong association with CL / CP. A trissomia 13 (Também conhecida como síndrome de Patau) está associada a três cópias do cromossoma 13, ou translocações Robertsonianas desequilibradas envolvendo o cromossoma 13. Bebês nascidos com esta condição normalmente morrem no período neonatal. As características clínicas incluem fissura labial e palato, atraso de crescimento, malformações nervosas centrais graves (incluindo holoprosencefalia), microcefalia, microptalmia, coloboma da íris, ausência dos olhos, orelhas malformadas, polidactilia, punhos cerrados, pés de fundo rochoso, defeitos cardíacos congênitos e defeitos urogenitais. Fissuras na linha média (caso contrário muito raras) podem ser observadas na trisomia 13 devido ao risco de defeitos na linha média, incluindo a holoprosencefalia. A trissomia 18 (Também conhecida como síndrome de Edwards) é tipicamente devido a três cópias distintas do cromossomo 18, e está associada a resultados pós-natais pobres. As características clínicas incluem fissura labial e palato, incapacidade intelectual, incapacidade de prosperar, doença cardíaca congênita, hipertonia, micrognathia, esterno curto, orelhas malformadas de baixo conjunto, mãos apertadas, pés de fundo rocker, e unhas hipoplásticas, entre outros. Trissomia 13 e 18 pode ser facilmente confirmada ou descartada através da análise cromossômica (cariotipagem).As síndromes de microdeleção tipicamente envolvem a eliminação de parte de um cromossomo. Estas supressões podem ser muito pequenas para serem detectadas por cariotipagem padrão e podem exigir a detecção de peixes (hibridização por fluorescência in situ) ou tecnologia de microarray. Uma síndrome de microdeleção bem conhecida associada à cleft palate é a síndrome de deleção 22q11.2 (também conhecida como síndrome de Digeorge/Velocardiofacial). Anormalidades palatais incluindo incompetência velofaríngea, fissuras submucosas, uvula bifid, e palato de fissura são vistas em 69% dos indivíduos com deleção 22q11.2, e podem ser parte da sequência Pierre Robin. Outros achados clínicos incluem doença cardíaca congênita, perda de audição, características dismórficas, deficiência imunológica, hipocalcemia, anomalias renais, problemas de alimentação, anomalias esqueléticas e distúrbios psiquiátricos. Aproximadamente 10% dos casos de síndrome de deleção 22q11.2 são considerados familiares. A exclusão segrega de forma autossômica dominante.(6) a síndrome Wolf-Hirschhorn, que se deve a uma deleção no braço curto do cromossoma 4, está também associada a fissuras orais (em 25% a 50% dos indivíduos afetados). Característica características faciais (incluindo proeminentes glabelar levando ao “grego-guerreiro de capacete aparência”), doença cardíaca congênita, deficiência mental, convulsões, insuficiência de crescimento, micrognatia, preauricular tags ou poços, e hypodontia também pode ser visto como parte da condição.(7)
distúrbios de gene único com fissuras orais incluem síndrome de Stickler, síndrome de Treacher Collins, e síndrome de Van der Woude, entre muitos outros. A síndrome de Stickler é uma doença de colagénio com hereditariedade autossómica dominante e, menos frequentemente, autossómica recessiva. Características comuns incluem fenda palatina (visto como parte de Pierre Robin sequência ou sem micrognatia), perda auditiva neurossensorial e condutor), esquelético resultados (início precoce da artrite, spondyloepiphyseal displasia), anomalias oculares (alta miopia, vítreo anomalias) e característica características faciais (com o subdesenvolvimento da maxila e ponte nasal, face média retrusion). Os testes genéticos para a síndrome de Stickler podem ser complexos, como mutações em pelo menos seis genes foram descritos em indivíduos afetados. Aproximadamente 90% dos pacientes com síndrome de Stickler têm mutações no gene COL2A1 e têm uma forma autossômica dominante da condição.(8) a síndrome de Treacher Collins é uma condição autossómica dominante caracterizada por fenda palatina com ou sem fenda labial em 28% dos indivíduos afetados. Outras anormalidades incluem hipoplasia dos ossos zigomáticos e mandíbula, anomalias do ouvido externo, coloboma da pálpebra inferior, perda auditiva condutiva, ausência de pestanas inferiores, deslocamento do cabelo preauricular nas bochechas, estenose ou atresia coanal. O diagnóstico da síndrome de Treacher Collins é baseado em achados clínicos e radiográficos. Foram descritas mutações em pelo menos três genes, com mutações no TCOF1 observadas em 78% a 93% dos doentes.(9) a síndrome de Van der Woude é caracterizada pela presença de fístulas paramedianas de lábios inferiores (pits) congênitas, geralmente bilaterais, ou por vezes pequenas montadas com um trato sinusal que conduz a partir de uma glândula mucosa do lábio, e fissuras orais (incluindo CL/CP e CP). Van der Woude é uma condição autossómica dominante associada a mutações no gene IRF6 (10). O teste para condições de gene único ou multi-gene requer a análise direta do gene por sequenciação e/ou análise de exclusão/duplicação (como o MLPA).
dado que as síndromes genéticas com fissura labial e palato podem ser associadas a aneuploidias, microdeletções cromossómicas/microduplicações, ou distúrbios do gene único, o teste genético pode ser um processo complicado. Uma história médica completa, um pedigree de três gerações, uma história de Gravidez, e um exame de dismorfologia por um geneticista clínico podem esclarecer o quadro clínico e permitir testes genéticos específicos. Novas tecnologias, incluindo microarray, permitirão a identificação de pequenas microdeletions e microduplicações anteriormente falhadas pela caryotipagem padrão. Infelizmente, esta técnica também leva à identificação de deleções e duplicações de significado clínico desconhecido, complicando o processo de aconselhamento genético. O teste para doenças de um único gene ou doenças mendelianas requer a disponibilidade clínica de testes genéticos para o gene desejado. Também pode ser caro se não for coberto pelo seguro médico. Novas tecnologias como sequenciamento de próxima geração, sequenciamento de exome ou sequenciamento do genoma (conhecidos coletivamente como testes genômicos) tornaram-se clinicamente disponíveis. Ao analisar centenas a milhares de genes simultaneamente, estes testes aumentam significativamente o poder de diagnóstico e rendimento. Em comparação com outras técnicas, estes testes podem fornecer uma resposta mais rápida e de forma mais eficiente em termos de custos. No campo da investigação, a sequenciação do exome e do genoma levou à identificação de novos genes, bem como à expansão das características clínicas e do espectro para mutações genéticas. Tal como acontece com a tecnologia microarray, os testes genômicos podem detectar síndromes que não estão relacionados com a apresentação do paciente e/ou a razão para o teste. Dadas as complexidades inerentes aos ensaios genéticos, é necessário o consentimento informado.
conclusão
embora a fenda labial e palato seja uma anomalia isolada na maioria dos casos, há uma forte associação entre fissuras orais e outras anomalias e síndromes genéticas. Uma avaliação genética por um geneticista clínico e um conselheiro genético é essencial para a orientação antecipada e para determinar os riscos para a recorrência. Os ensaios genéticos, que exigem consentimento informado, podem ser coordenados e interpretados durante uma avaliação genética.Anya Revah, MS, é a conselheira genética sênior da Divisão de Genética Médica no Maimonides Infants and Children’s Hospital em Brooklyn, Nova Iorque. Ela também é membro ativo do Centro Médico de Maimonides e da equipe multidisciplinar do Kings County Hospital Cleft Lip e Palate. Ela tem um Mestrado em Ciência em aconselhamento genético da Universidade de Boston, em Boston, Massachusetts.
1. Sadler TW. A embriologia médica de Langman. Nona Edição. Pages 390-395.
2. Parker SE, Mai CT, Canfield MA, Rickard R, Wang Y, Meyer RE, Anderson P, Mason CA, Collins js, Kirby RS, Correa A. For the National Birth Defects Prevention Network. Estimativas atualizadas da prevalência Nacional de natalidade para defeitos de nascença selecionados nos Estados Unidos. 2004-2006. Birth Defects Research (Part A): Clinical and Molecular Teratology 2010;88:1008-1016.
3. Fraser FC. A genética da fenda labial e fenda palatina. Manha. J. Hum. Genet. 1970;22: 336–352.
4. Van Rooij IA, Ocke MC, et al. A ingestão de folato Periconceptual por suplemento e a ingestão de alimentos reduzem o risco de fenda labial não-sindrómica, com ou sem fenda palatina. Prev Med 2004; 39: 689-694.
5. Tan TY. Kilpatrick N, Farlie PG. Perspectivas genéticas e de desenvolvimento sobre a sequência de Pierre Robin. Manha. J. Med. Genet. 2013; 163C: 295-305.
6. McDonald-McGinn DM, Emanuel BS, Zackai EH. Síndrome de deleção 22q11.2. Setembro. 23, 1999. . In: Pagon RA, Adam MP, Ardinger HH, et al., Editor. Generevews . Seattle( WA): University of Washington, Seattle; 1993-2014. Disponível a partir de: http://www.ncbi.nlm.nih.gov/books/NBK1523/.
7. Battaglia A, Carey JC, South ST, et al. Síndrome Wolf-Hirschhorn. Abril. 29, 2002. . In: Pagon RA, Adam MP, Ardinger HH, et al., Editor. Generevews . Seattle( WA): University of Washington, Seattle; 1993-2014. Disponível em: http://www.ncbi.nlm.nih.gov/books/NBK1183/.
8. Robin NH, Moran RT, síndrome de Ala-Kokko L. Stickler. Junho. 9, 2000. . In: Pagon RA, Adam MP, Ardinger HH, et al., Editor. Generevews . Seattle( WA): University of Washington, Seattle; 1993-2014. Disponível em: http://www.ncbi.nlm.nih.gov/books/NBK1302/.
9. Katsanis SH, Jabs EW. Síndrome De Treacher Collins. Julho. 20, 2004. . In: Pagon RA, Adam MP, Ardinger HH, et al., Editor. Generevews . Seattle (WA): University of Washington, Seattle; 1993-2014. Disponível em: http://www.ncbi.nlm.nih.gov/books/NBK1532/.
10. Schutte BC, Saal HM, Goudy S, et al. Doenças relacionadas com o IRF6. Outubro. 30, 2003. . In: Pagon RA, Adam MP, Ardinger HH, et al., Editor. Generevews . Seattle( WA): University of Washington, Seattle; 1993-2014. Disponível em: http://www.ncbi.nlm.nih.gov/books/NBK1407/.