Autossômica Recessiva Ictiose Congênita | Actas Dermo-Sifiliográficas
Introdução
O mais recente consenso classificação de ictiose diferencia entre 2 formas principais: o nonsyndromic formas, que se apresentam com as manifestações cutâneas, e o sindrômico formas, que se apresentam com manifestações em outros órgãos, bem como (Tabela 1).1 entre as formas não-sindrômicas, quatro grupos são identificados: ictioses comuns, ictioses autossômicas recessivas congênitas (ARCIs), ictioses queratinopáticas e outras ictioses menos comuns.Tradicionalmente, o grupo de ARCIs era dividido em 2 transtornos, ictiose lamelar (LI) e ictiosia congênita eritroderma (CIE). Na nova classificação, ictiose arlequim (HI) foi adicionada a este grupo1 porque inactivação de mutações no ABCA12 gene têm sido identificados como responsáveis por este transtorno,2,3, enquanto que absurdo mutações no mesmo gene podem dar origem à LI4 ou CIE5,6 fenótipo. Outras variantes menos comuns incluídas no grupo de ARCIs são colodion baby (SHCB), acral SHCB, e ictiose de fatos de banho.7-9
classificação consensual baseada nas características clínicas da Ictiosis1.
| Nonddromic Formas | Sindrômico Formas |
| Comum IchthyosesIchthyosis vulgarisRecessive x-linked ictiose (nonsyndromic )ajimajor formsHarlequin ichthyosisLamellar ichthyosisCongenital ichthyosiform erythrodermaMinor formsSelf-cura do colódio babyAcral de auto-cura do colódio babyBathing terno ichthyosisKeratinopathic IchthyosesMajor formsEpidermolytic Ichthyosissuperficial epidermolytic ichthyosisminor formsannular epidermolytic ichthyosiscurth-Macklin ichthyosisautosomal recessiva epidermolytic ichthyosisEpidermolytic nevusOther FormsLoricrin keratodermaErythrokeratodermia vararabilispeeling pele syndromeCongenital reticular ichthyosiform erythrodermaKLICK síndrome | Sindrômico X-linked Ichthyosrecessive x-linked ictiose (sindrômico)Ictiose follicularis, alopecia, e fotofobia (IFAP) syndromeconradi-Scammann-happle (síndrome de condrodisplasia punctata tipo 2)sindrômico autossômica ichthyosisskin disordersnetherton syndromeichthyosis-hypothrichosis syndromeichthyosis-Colangite Esclerosante syndrometrichothystrophyneurological Disorderssjögren-Larsson syndromeRefsum diseaseMEDNIK syndromeFatal disease courseGaucher disease, type 2Multiple sulfatase deficiencyCEDNIK syndromeARC syndromeOther associated signsKID syndromeChanarin-Dorfman syndromeIchthyosis prematurity syndrome |
Abbreviations: ARC, arthrogryposis–renal dysfunction–cholestasis; ARCI, autosomal recessive congenital ichthyosis; CEDNIK, cerebral dysgenesis, neuropathy, ichthyosis, and palmoplantar keratoderma; KID, keratitis ichthyosis deafness; KLICK, keratosis linearis with ichthyosis congenital and sclerosing keratoderma; MEDNIK, atraso mental, enteropatia, surdez, neuropatia periférica, ictiose, queratoderma.
os dados disponíveis sobre a epidemiologia do ARCIs são limitados. Nos Estados Unidos, estimou-se a prevalência no nascimento de 1 por 100 000 habitantes para LI e de 1 por 200 000 habitantes para CIE. Outros estudos relataram uma prevalência combinada de LI e CIE de 1 por 200 000 a 300 000 habitantes.10,11 em alguns países, como a noruega, a prevalência estimada é maior (1 por 91 000) devido a mutações fundadoras.12 A constatação de 1 ou várias mutações recorrentes em uma população pode ser porque a mutação ocorreu em um determinado ponto na história e, em seguida, foi passado de geração para geração (fundador mutação) ou porque a região do genoma onde a mutação é encontrada possui uma sequência de DNA suscetíveis a mutação (mutation hotspot). Em Espanha, a prevalência estimada de ARCI é de 1 por 138 000 na população em geral e de 1 por 61 700 entre crianças com menos de 10 anos de idade.13 em certas regiões de Espanha, a prevalência pode ser ainda mais elevada. Na costa galega, por exemplo, foi relatada uma prevalência de 1 por 33 000, devido também a um efeito fundador.14
ictiose lamelar e ictiose congênita características Eritrodermaclínicas
embora se tenha pensado originalmente que LI e CIE eram entidades diferentes, houve relatos de pacientes com manifestações clínicas intermediárias e ambas as condições podem ser causadas por mutações no mesmo gene.15,16 além disso, pacientes com a mesma mutação, mesmo dentro da mesma família, podem desenvolver fenótipos diferentes.12,15
a maioria dos doentes nascem envoltos numa membrana de colódio que desaparece progressivamente durante as primeiras semanas de vida e é substituído pelo fenótipo definitivo (Fig. 1A). A hipohidrose, a intolerância ao calor grave e a distrofia das unhas são frequentemente observadas tanto na LI como na CIE.17-19 pacientes com LI geralmente têm manifestações clínicas mais graves do que aqueles com CIE. Eles têm grandes escamas plateliais, muitas vezes de cor escura, cobrindo toda a área da superfície do corpo. O eritroderma ou está ausente ou é mínimo. Tais pacientes geralmente têm ectropião e, por vezes, eclábio, hipoplasia da cartilagem articular e nasal, cicatrizando alopecia, especialmente na borda do couro cabeludo, e queratoderma palmoplantar (Fig. 1B e C). A CIE é caracterizada pela presença de eritroderma e escamas esbranquiçados (Fig. 2). Alguns doentes têm eritema marcado e escala generalizada. As escamas podem ser grandes e de cor escura, particularmente nas superfícies extensoras das pernas. Em casos menos graves, o eritema é ligeiro e a escala é óptima.

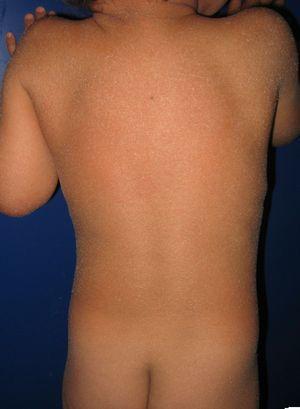
doente com eritroderma ictiosiforme congénito e mutações no gene ALOXE3. Eritema ligeiro e descamação generalizada do furfuráceo esbranquiçado.
as alterações histopatológicas
histopatológicas não fornecem um diagnóstico. Em LI, hiperqueratose ortoceratótica massiva é observada, geralmente com o dobro da extensão como em CIE. A epiderme é acantótica e ocasionalmente assume uma aparência psoríase. A taxa de proliferação celular é normal ou ligeiramente elevada.17-19 doentes com CIE têm hiperqueratose menos marcada, com paraceratose focal ou extensa, uma camada granular normal ou espessada, e acantose mais pronunciada. A rotação epidérmica é aumentada.17-19
ultra-estrutura
embora uma estreita correlação entre descobertas moleculares, clínicas e ultra-estruturais ainda não tenha sido encontrada, microscopia eletrônica pode, no entanto, ser útil para descartar outras formas de ictiose e para orientar análises genéticas em alguns casos. Foram descritos quatro tipos de ictiose congénita (Tabela 2).
classificação ultra-estrutural de Ictioses congênitas.
| Tipo | Característica Principal | Outros Recursos | Mutações | Manifestações Clínicas |
| 1 | a Ausência de marcadores ultra-estrutural de ictiose do tipo 2, 3, e 4 | gotículas de Lipídios ou anéis no estrato córneo (o mais frequente)Pequeno keratohyalin granulesVesicular ou lobular membrana de revestimento de grânulos | TGM1 (33.3%)ALOX12B (2 casos) | CIE |
| 2 | Colesterol fissuras no estrato córneo | a Ausência ou enfraquecimento do cornified envelopeSmall keratohyalin granulesLipid gotas | TGM1 (89-100%) | LI |
| 3 | Laminado membraneous estruturas no stratum granulosum e/ou do estrato córneo. | Anormal da membrana de revestimento granulesLipid dropletsFoci de destaque juxtanuclear vacúolos na camada granular | NIPAL4 (93%) | CIE (o mais frequente)LI |
| 4 | Trilamellar membrana pacotes que preencher algumas células no stratum granulosum e/ou estrato córneo | Anormal da membrana de revestimento de grânulos | FTAP4 | Ictiose prematuridade, síndrome de (100%) |
Abreviaturas: (CIE), congênitas ichthyosiform erythroderma; LI, ictiose lamelar.
ictiose congênita tipo 1
ictiose congênita tipo 1 é caracterizada pela ausência de marcadores Ultra-estruturais para ictiose tipos 2, 3 e 4. Portanto, o diagnóstico é geralmente feito apenas quando os outros tipos foram excluídos. A descoberta mais frequente é a presença de gotículas lipídicas ou anéis no estrato córneo (Fig. 3A).20 estas gotículas lipídicas não são uma característica constante ou específica a este tipo particular,uma vez que não estão presentes em todos os casos, 20 e podem estar presentes em outros tipos de ictiose.21,22 clinicamente, a maioria dos pacientes apresenta manifestações de CIE.12,20 um terço dos pacientes tem mutações no gene TGM1.Este tipo ultra-estrutural também foi identificado em associação com mutações no gene ALOX12B.23,24

imagens de microscópio electrónico. A, ictiose congênita tipo 1, mostrando gotículas lipídicas no estrato córneo e ausência de marcadores Ultra-estruturais dos outros tipos de ictiose. B, ictiose congênita tipo 2, caracterizada pela presença de fissuras de colesterol (seta) nos córneos.
Ictiose Congênita Tipo 2
ictiose Congênita tipo 2 é caracterizado por colesterol fissuras no estrato córneo (Fig. 3B).21 tais fissuras são um achado constante neste tipo de ictiose, e pode ser detectado em diferentes biópsias no mesmo paciente; o tratamento com retinóides orais não tem impacto nestas fissuras.Foram também observados 12,25 agregados com densidade de electrões em corneócitos em alguns doentes com actividade deficiente da TGase 1.26-28 clinicamente, a maioria dos doentes apresenta manifestações graves de CIE.Este tipo ultra-estrutural está fortemente associado com mutações no gene TGM1.12,16
ictiose congénita Tipo 3
ictiose congénita tipo 3 é caracterizada por estruturas membranosas lamelares no estrato granuloso e/ou estrato córneo. Estas estruturas são dispostas em tiras em torno de um espaço vazio perto do núcleo.22,29-31 as manifestações clínicas deste tipo são diferentes das outras; o início da ictiose é variável, a descamação e o eritema podem ser irregulares ou generalizados, e as flexões em particular são afetadas. As mutações no gene NIPAL4 são responsáveis por 93% das ictioses tipo 3.32
ictiose congénita Tipo 4
caracteristicamente, na ictiose congénita tipo 4, algumas células do estrato granuloso e do estrato córneo estão cheias de embalagens de membrana trilamelar.Estes achados são patognômicos para a síndrome de prematuridade da ictiose, uma condição atualmente considerada como uma forma sindrômica de ictiose.34,35
estudos moleculares
em termos genéticos, os ARCIs são muito heterogéneos. O gene TGM1 está associado com a maioria dos casos, mas foram notificadas mutações em 5 outros genes (ALOX12B, ALOXE3, NIPAL4, CYP4F22 e ABCA12). Fischer et al.36 estudou 520 famílias com ARCI e identificadas mutações em pelo menos 1 destes genes em 78% dos casos (TGM1 em 32%, NIPAL4 em 16%, ALOX12B em 12%, CYP4F22 em 8%, ALOXE3 em 5%, e ABCA12 em 5%). Num outro estudo de 250 doentes com ICAR de origens diferentes, 38% tinham mutações TGM1, 6, 8% tinham mutações ALOXE3 e 6, 8% tinham mutações ALOX12B.37 na Galiza, identificamos mutações nos genes TGM1, ALOX12B, ALOXE3, NIPAL4 e CYP4F22 em 75% das famílias estudadas, mas a distribuição das mutações foi diferente.14 o gene TGM1 sofreu uma mutação em 68.7% dos casos enquanto o gene ALOXE3 sofreu mutação em apenas 1 doente. Não detectamos mutações em nenhum dos outros 3 genes estudados.
TGM1
o gene TGM1 está localizado no cromossomo 14q11. 2 e tem 15 exons (GenBank NM-000359.2). Codifica a enzima TGase 1, que é uma das 3 enzimas TGase encontradas na epiderme.38 esta enzima participa na formação do envelope cornificado por catalizar a ligação cruzada dependente do cálcio de várias proteínas, tais como a involucrina, a loricrina e as proteínas ricas em prolina.39,40 também catalisa a ligação de ??- hidroxiceramidas na camada exterior do envelope cornificado com proteínas na camada interior.41,42 em doentes com mutações TGM1, o envelope cornificado está em falta e a actividade da TGase 1 é reduzida ou inexistente.43-47
desde 1995,quando este gene foi identificado como responsável por alguns casos de ICAR, foram notificadas 48-50 mais de 110 mutações em doentes de origens diferentes. Mutações em TGM1 são a causa mais comum de ARCI.36,37 esta mutação foi encontrada em 55% dos casos nos Estados Unidos e em 84% dos casos na Noruega.12,51 a mutação mais frequente é C.877-2A> G, que foi encontrado em 34% dos alelos mutados notificados até à data.A elevada frequência desta mutação em países como os Estados Unidos e a Noruega deve-se a um efeito fundador.12.53 a segunda mutação mais frequente é P. Arg142His. Esta e outras mutações semelhantes foram relatadas em países como Egito, Alemanha, Finlândia e Estados Unidos, 15,49-51,54-56 e parece que estas são mutações de hotspot.A mutação P. Arg307Trp é frequente na população japonesa.5 na Galiza, A P.Arg760X, C. 1223_1227delACACA e C.984+1G> a mutações no TGM1 foram identificadas em 81, 82% das famílias com mutações neste gene, sugerindo um efeito fundador.14 a confirmação desta hipótese foi obtida pelo estudo haplótipo (trabalho ainda não publicado).
mutações TGM1 são responsáveis pela maioria dos casos de LI15,27,44,46,56,58-63 e por uma pequena percentagem de casos de CIE.43,47,64,65 tais mutações também podem dar origem a outras formas de ARCI, tais como SHCB, acral SHCB, e ictiose do fato de banho.
muitos estudos tentaram demonstrar associações genótipo-fenótipo entre mutações em TGM1 e resultados Ultra-estruturais ou clínicos, mas não foi observada qualquer correlação significativa até à data.15. 16. 53 em geral, os doentes com mutações no gene TGM1 são mais gravemente afectados do que os doentes sem tais mutações. Num estudo com 83 doentes com ARCI na Suécia e na Estónia, a presença de ectropião e de bebé colodiónico foi associada a mutações TGM1, tendo sido observada uma taxa mais elevada de eritema em doentes sem mutações neste gene.Outro estudo mostrou que o tipo de escala é a principal diferença entre portadores e não-Carregadores de mutações TGM1, ao descobrir que todos os pacientes com mutações neste gene tinham escala lamelar enquanto 80% dos que não tinham mutações TGM1 tinham escala fina.Além disso, verificou-se que as mutações truncantes estão mais frequentemente associadas a hipohidrose e perturbações da sudação do que as mutações de missense.51 na população norte-americana, um modelo baseado na presença de certas características clínicas prevê que os doentes que nascem como bebés colódios e têm distúrbios oculares e/ou alopecia têm 4 vezes mais probabilidade de ter mutações TGM1.51
ALOXE3 e ALOX12B
O ALOXE3 e ALOX12B genes estão localizados no cromossomo 17p13.1.67, Eles tem uma estrutura semelhante com 15 éxons que codificam a epiderme LOXs eLOX-3 e 12R-LOX.68,69 o fato de que eles são predominantemente expressos nas camadas supra-basais da epiderme suporta seu papel em fases avançadas de diferenciação epidérmica, com participação no processamento de corpos lamelares.24,70 estas enzimas actuam em degraus adjacentes na Via da hepoxilina(Fig. 4). O 12R-LOX transforma o ácido araquidónico em ácido 12R-hidroxieicosatetraenóico,enquanto o eLOX-3 converte este produto num isómero epoxialcohol69, 71 da família hepoxilina A3.O produto da hepoxilina é instável e hidrolisado nas células para um derivado Tri-hidroxi específico (trioxilina). Embora o papel exacto dos produtos da via hepoxilina não seja conhecido, especula-se que estes podem participar na formação de lípidos intercelulares do estrato córneo ou actuar como sinais para induzir a diferenciação queratinocitária.
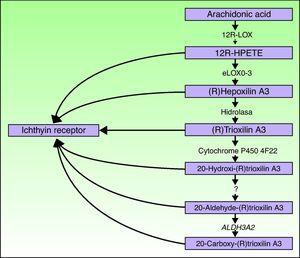
os genes ALOX12B e ALOXE3 foram identificados pela primeira vez em 2002.73,74 desde então, mais de 30 mutações nos gene23,24,37,75-77 e aproximadamente 10 nos gene37,74,75. Estas mutações são responsáveis por 14% a 17% de ARCIs36,37 e 72.2% da SHCBs.23,78,79 a relação causal entre estas mutações e fenótipo foi confirmada através da demonstração de que a actividade catalítica do LOX epidérmico foi totalmente abolida em doentes com estas mutações 75,80 e através da utilização de modelos animais que reproduziam o fenótipo ictiosiforme observado em seres humanos.81-83 ambos os genes são responsáveis por uma percentagem semelhante de casos ARCI. No entanto, o intervalo de diferentes mutações no gene ALOXE3 é limitado, devido à predominância de 2 mutações, P.Arg234X E P.Pro630Leu, que parecem corresponder a pontos quentes.37, 74, 75
os doentes com mutações nos genes ALOXE3 e ALOX12B apresentam geralmente um fenótipo da CIE.74,75,77 a gravidade da escala é leve ou moderada, e as escamas têm uma cor esbranquiçada ou castanho-claro. Pode também estar presente eritema. 76% dos pacientes nascem como bebês de colódios e 88% têm problemas de transpiração.37 doentes com mutações no gene ALOX12B apresentam uma descamação mais limitada e esbranquiçada em comparação com portadores de mutações no gene ALOXE3. Nestes casos, as escamas são acastanhadas e aderentes. A presença de eritema, hiperqueratose palmoplantar e acentuação das dobras palmoplantar também estão associadas a mutações ALOX12B.O gene NIPAL4, também conhecido como gene ichthyin, está localizado no cromossomo 5q33. Tem 6 exões que codificam uma proteína com vários domínios transmembranares de função desconhecida.Foi colocada a hipótese de que o produto proteico participa na mesma via metabólica que o LOX e pode actuar como receptor para as trioxilinas A3 e B3 ou para outros metabolitos da via metabólica da hepoxilina.84 seria assim implicada na formação de corpos lamelares ou no seu transporte para o espaço extracelular.32 em apoio a isso são duas observações. Em primeiro lugar, em 93% dos casos, mutações neste gene estão associadas a um padrão ultra-estrutural de ictiose congênita tipo 3, caracterizada por anormalidades nos corpos lamelares e a presença de membranas perinucleares alongadas no estrato granuloso.32 em segundo lugar, NIPAL4 é expresso essencialmente no estrato granuloso da epiderme, onde os corpos lamelares estão presentes.85
desde a descoberta do gene NIPAL4 em 2004, apenas 9 mutações foram relatadas em pacientes de países mediterrânicos (Argélia, Turquia e Síria),84 países escandinavos,32 Paquistão,85 Ilhas Faroé,32 e América do Sul.84
o espectro clínico de doentes com mutações neste gene é amplo, mesmo entre membros da mesma família. Entre 3,7% 32 e 60% 84 nascem como bebés de colódio. Quando a membrana de colódio desaparece, a maioria dos pacientes desenvolve as manifestações de CIE, com escamas esbranquiçadas finas em uma base eritematosa na face e tronco e maiores, escamas acastanhadas no pescoço, nádegas e pernas.Xerose marcada, placas hiperqueratóticas reticulares acastanhadas generalizadas que parecem acentuadas nas dobras da pele, e discromia facial podem estar presentes.32,85 além disso, queratoderma palmoplantar é uma descoberta frequente, juntamente com contraturas ocasionais de dedos e unhas curvadas de dedos. Alguns estudos relataram achados mais típicos de LI.32,85 foi notificada a presença de sinais e sintomas de dermatite atópica em alguns doentes, embora não tenham sido detectadas mutações no gene FLG em nenhum destes casos.85
CYP4F22
O FLJ39501 ou CYP4F22 gene está localizado no cromossoma 19p13.12.86 12 exons87 e codifica uma citocromo P450, família 4, subfamília F, polipeptídeo 2, homóloga de leucotrieno B4 – ω-hydroxylase (CYP4F2). A reação catalisada pelo produto de FLJ39501 na pele e os substratos dessa reação podem ser deduzidos por analogia com seus homólogos conhecidos CYP4F2 e CYP4F3.Foi colocada a hipótese de que o CYP4F2 e o CYP4F3 participam na Via hepoxilina catalisando a conversão da trioxilina A3 em 20-hidroxi-(R)trioxilina A387 e que o produto final desta via, 20-carboxi-trioxilina A3, pode ter um efeito regulador biológico fundamental na pele.89
até à data, apenas 8 mutações deste gene foram notificadas em 12 famílias consanguíneas de países mediterrânicos 87 e em 1 família de origem israelita.62
nas famílias reportadas por Lefèvre et al., 87 a maioria dos doentes tinha um fenótipo da CIE à nascença e este evoluiu subsequentemente para LI. os doentes nasceram geralmente com eritroderma marcado, embora sem qualquer membrana colodiónica. À medida que envelheciam, desenvolveram uma escala generalizada de cinza-branco, que era mais marcada na região periumbilar, nas nádegas, e na parte inferior do corpo. A hiperlinearidade das palmas e solas e a descamação no couro cabeludo, em tempos de tipo pitiriasiforme, eram frequentes.87 em outra família, os três membros afetados nasceram como bebês de colídio e desenvolveram eritroderma intensa, descamação generalizada e keratoderma palmoplantar.62
ABCA12
em 2003, foi relatado que o gene ABCA12 era responsável por alguns casos de LI e foi mapeado para o cromossoma 2q34.4 foi subsequentemente confirmado que as mutações neste gene eram também responsáveis pela HI.2,3ABCA12 codifica 53 éxons, e pertence a uma família de transportadores ABC, que se ligam de trifosfato de adenosina, facilitando o transporte de várias moléculas através da membrana celular.90 os membros da subfamília ABCA estão todos implicados no transporte de lípidos.A insuficiência da função ABCA12 causa distúrbios do transporte lipídico nos corpos lamelares, conduzindo assim a uma diminuição dos níveis lipídicos intercelulares no estrato córneo.Estudos de infra-estruturas demonstraram que o ABCA12 está localizado em corpos lamelares associados a glicosilceramidas.91ABCA12 mutações têm sido associadas a distúrbios na distribuição e o transporte de glycosylceramides e com a diminuição dos níveis de hydroxyceramides, um dos principais componentes da barreira lipídica nos espaços intercelulares.A hiperqueratose massiva que ocorre nestes pacientes pode ser uma resposta compensatória a uma barreira lipídica deficiente.94 também pode ser devido à falta de descamação dos córneos,93 que pode ser causada por defeitos no transporte de certas proteases, tais como calicreina 5 e catepsina D, resultantes de distúrbios nos corpos lamelares.Modelos murinos e estudos in vitro sugerem que as mutações ABCA12 também têm um efeito na diferenciação epidérmica.95-97
até à data, foram notificadas mais de 50 mutações no gene ABCA12 em doentes com ARCI de África, Europa, Paquistão e Japão. As mutações mais frequentes são P. Val244SerfsTer28,2,98,99 identificadas em populações paquistanesas e indianas, e P. Asn1380Ser,4 identificadas em famílias africanas. Em ambos os casos, estas podem ser mutações fundadoras.
a extensão das mutações ABCA12 está relacionada com o fenótipo, com mutações associadas a perda completa de função conduzindo ao fenótipo HI.2,3,98-102 pelo contrário, em LI e CIE, a maioria das mutações são o missense, e têm um efeito menos grave na função proteica.4-6, 103 as mutações subjacentes ao fenótipo LI parecem estar concentradas na primeira região cassete de ligação trifosfato de adenosina.Clinicamente, os doentes com CIE e mutações no gene ABCA12 têm escalas de tamanho médio que são um pouco maiores do que as normalmente observadas em doentes com este fenótipo.
a ictiose da Arlequina
HI ou o feto da Arlequina é uma forma grave e geralmente fatal de ictiose. As crianças são geralmente prematuras com extensas placas hiperkeratóticas brilhantes, separadas por fissuras profundas, que cobrem todo o tegumento e formam padrões geométricos reminiscentes de roupas usadas por Arlequins, dando assim à condição o seu nome. A aperto da pele leva a uma eversão marcada das pálpebras e lábios, desenvolvimento rudimentar da cartilagem articular e nasal e, ocasionalmente, microcefalia. As crianças raramente têm pestanas ou sobrancelhas, embora o cabelo no couro cabeludo pode ser conservado. As mãos e os pés estão inchados e edematosos, e muitas vezes cobertos por uma camada semelhante a uma luva. Podem ter contracções nos dedos.
para estes doentes, o risco de morte durante o período neonatal é muito elevado.104 ventilação pulmonar está comprometida; perda de água transepidermal leva à desidratação, desequilíbrio hidrelétrico e instabilidade térmica; e o risco de infecções é aumentado. A aperto Facial e o eclabium dificultam a sucção e, portanto, a alimentação, com o correspondente agravamento da desidratação. Os recém-nascidos com esta condição raramente viveram mais algumas semanas. Nos últimos anos, no entanto, as chances de sobrevivência a longo prazo aumentaram notavelmente, essencialmente devido à administração de retinóides sistêmicos e progresso nos cuidados intensivos neonatais.Num estudo recente, 83% dos doentes tratados com retinóides orais sobreviveram comparativamente a 24% dos doentes não tratados. A maioria das mortes ocorreu nos primeiros 3 dias de vida, mas o tratamento só foi iniciado depois disso em muitos dos sobreviventes.Isto sugere que muitas destas mortes precoces teriam ocorrido independentemente do tratamento retinóide.
as crianças que sobrevivem ao período neonatal geralmente desenvolvem CIE grave.A natureza e localização das mutações no gene ABCA12 e a extensão da perda da função do transportador podem determinar o prognóstico.3.922.107 pacientes que conservam um certo grau de atividade proteica, embora mínima, podem ter uma melhor chance de sobreviver. Portadores de mutações homozigóticas têm uma taxa de mortalidade mais elevada.104
a principal característica histológica do HI é a presença de um estrato córneo ortoceratótico extremamente espesso e compacto. Os folículos capilares e os canais de suor têm plugs107,108 proeminentes hiperkeratóticos e têm corpos lamelares anormais ou ausentes, inclusões lipídicas, ou restos de organelas ou núcleos nos córneos, e ausência de lípidos intercelulares no estudo ultra-estrutural.108.109 os folículos capilares apresentam uma concentração marcada de material queratótico, que é uma característica diagnóstica do HI utilizado para o diagnóstico pré-natal.
até à data, a taxa de detecção de mutações no gene ABCA12 em doentes com IH é próxima de 100%, o que parece ser uma condição geneticamente homogénea.
Collodion Baby and Self-healing Collodion Baby
Collodion babies are usually born premature and perinatal morbity and mortality are increased. No nascimento, o recém-nascido é coberto por uma membrana transparente ensinada brilhante reminiscente do Invólucro celofano(Fig. 5). Os bebês têm ectropion, eclabium, e hipoplasia da cartilagem nasal e articular. A sucção e a ventilação pulmonar podem ser impedidas110 e a perda transepidérmica de água e o risco de infecções são aumentados.110,111

Colodion baby that subsequently progressed to a lamellar ichthyosis phenotype.
Collodion baby é a apresentação habitual para HI e CIE. Autossómica dominante LI,112,113 Sjögren-Larsson síndrome,110 trichothyodystrophy,114 juvenil, doença de Gaucher,110 neutro depósito de lipídios doença, Conradi-Hünermann-Happle síndrome de Hays-Poços de síndrome de ectodermal dysplasia115 também pode, ocasionalmente, apresentar como colódio bebê. A membrana desaparece espontaneamente em 10% a 24% dos recém-nascidos, dando lugar a uma pele completamente normal.110,116 no passado, estes casos foram descritos como LI do recém-nascido,117 mas não são referidos como SHCB.118 Alguns autores têm sugerido o termo auto-melhoria do colódio ictiose, porque muitos desses pacientes, quando retomada, mais tarde, na infância ou como adultos, têm um grau variável de anhidrosis e intolerância ao calor e leves sinais de ictiose, tais como xerosis e fina descamação, principalmente nas axilas e pescoço.78
nem a microscopia óptica nem as investigações Ultra-estruturais do bebé colódio são específicas. É, portanto, preferível adiar a biópsia da pele até que o fenótipo definitivo tenha se desenvolvido.Foram identificadas mutações
nos genes TGM1,7,119ALOXE3,78 e ALOX12B23,78,79 em doentes com HBC. As mutações de ALOX12B são as mais comuns. Numa série de 15 doentes escandinavos com SHCB, 67% apresentavam mutações no gene ALOX12B, 25% no gene ALOXE3 e 8, 3% no gene TGM1.78 mutações não foram encontradas em alguns pacientes, e assim outros genes também são susceptíveis de ser implicados. Tem havido especulações de que estas mutações reduzem a actividade enzimática no útero, mas não após o nascimento.7 no útero, onde a pressão hidrostática é elevada, a quelação por água Converte a enzima mutante numa conformação inactiva. Após o nascimento, quando a pressão diminui, a enzima regressa à sua forma activa e a sua actividade aumenta o suficiente para manter um fenótipo normal ou minimamente afectado.7
bebé de Colódios Acrais
embora o bebé de colódios afecte o corpo inteiro, foram notificados casos confinados às regiões acrais. In 1952, Finlay et al.120 relatou um caso de membrana de colódio que afetou apenas as mãos e pés e que seguiu um curso de auto-cura. Recentemente, um novo caso de acral SHCB foi relatado em associação com mutações do gene TGM1.Não se sabe por que razão estas lesões se restringem a regiões acrais, embora possam estar em funcionamento factores associados à regulação local-dependente da actividade enzimática.8
ictiose dos Fatos de banho
a ictiose dos fatos de banho foi pela primeira vez notificada como variante ARCI independente em 2005, embora tenham sido previamente notificados casos de ictiose com uma distribuição peculiar.121-123 foi detectado principalmente em pacientes de origem sul-africana, 9 embora também tenha sido relatado em indivíduos da Europa e países mediterrânicos.124 à nascença, os pacientes têm uma membrana de colódio generalizada que, em seguida, quebra para deixar a distribuição característica da escala. O tronco, região proximal dos braços, incluindo as axilas, o pescoço e o couro cabeludo são geralmente afetados, enquanto a parte central da face, membros, e a adrenal região são normalmente poupados.9 as escamas são grandes, lamelares e de cor escura. A descamação mais fina pode ocorrer no fossae popliteal e antecubital.124,125 as palmas das mãos e solas dos pés têm hiperqueratose difusa leve, enquanto as costas das mãos e pés não mostram envolvimento.
o estudo histopatológico da pele afectada mostra hiperqueratose marcada sem paraceratose, camadas granulares normais, acantose ligeira ou moderada e infiltração linfocítica ligeira na derme superior.9 observações de microscopia eletrônica são consistentes com ictiose congênita tipo 2 na maioria dos casos. A pele não envolvida não mostra quaisquer resultados anormais.124,125 em pele saudável, a actividade da TGase 1 é ligeiramente reduzida e geralmente localizada em áreas pericelulares. Na pele envolvida, a atividade enzimática é residual e anormalmente localizada no citoplasma.124 mutações
foram detectadas no gene TGM1 em todos os doentes com ictiose do fato de banho estudados até à data.119,124-126 a mutação mais comum é a P. Arg315Leu, que foi identificada na maioria dos pacientes Sul-africanos e pode ser uma mutação fundadora. Oji et al.124 sugeriu que a temperatura da pele poderia desempenhar um papel no desenvolvimento destas manifestações. Usando a termografia digital, os autores mostraram uma forte correlação entre a temperatura corporal e a descamação, sendo as áreas mais quentes do corpo as mais afetadas. Aufenvenne et al.Em doentes com ictiose do fato de banho verificou-se uma diminuição da temperatura óptima para a actividade do TGase 1. Esta diminuição não foi observada em controlos saudáveis ou em doentes com LI generalizada. esta diminuição na temperatura explicaria o fenótipo destes doentes. A temperatura óptima é de 37 ° C para a enzima normal, mas de 31°C para a enzima mutada.
tratamento
o principal objectivo do tratamento na ictiose é eliminar a escala e reduzir a xerose sem causar irritação excessiva (Tabela 3). Antes de decidir sobre o tratamento, aspectos como idade e sexo do paciente, tipo e gravidade da doença, e extensão e local das lesões devem ser levados em consideração.128
estratégia terapêutica em Ictioses congénitas Autossómicas recessivas.
| estratégia Terapêutica para o autossômica recessiva congênita ichthyoses | |
| Banho e mecânica, eliminação de escalas | Banho com bicarbonato de sódio ou amido de trigo, amido de milho, de arroz ou amido de; remoção mecânica das escamas (1 ou 2 vezes por dia) |
| tratamento tópico (sequencial) | hidratantes contendo ureia com queratinolíticos de propilenoglicol, ácidos α-hidroxi ou ureia combinados com neonatos de retinoidsina salicílica ácida e crianças pequenas, aplicar um veículo sem ingredientes activos. Evitar uréia, ácido salicílico e ácido lático, devido ao risco de absorção sistêmica |
| tratamento por via Oral | retinóides Orais (acitretina ou isotretinoin) |
| Outras medidas | acompanhamento de ectrópio pelo ophthalmologistRegular limpeza da orelha externa da orelha-de-garganta – -nariz specialistPhysiotherapy para evitar contraturas.Prevenção de actividades extenuantes numa hidroterapia de temperatura ambiente elevada |
recomenda-se a eliminação mecânica e de banhos de escamas
para doentes com ARCI, a fim de eliminar mecanicamente escamas e vestígios de hidratante. Isto é mais fácil se o paciente estiver imerso em água por 15 a 30 minutos. Alguns autores recomendam a adição de bicarbonato de sódio ao banho para desnaturar as queratinas e fazer a água alcalina, e assim facilitar a eliminação das escamas.129 outros produtos que podem ser adicionados incluem amido de trigo, amido de milho, ou amido de arroz. Os óleos de banho não são adequados, uma vez que podem conduzir à oclusão com subsequente risco de proliferação bacteriana e agravamento da termorregulação.
tratamento tópico
Hidratantes e agentes ceratolíticos tópicos são geralmente a primeira opção terapêutica. Melhoram a função da barreira cutânea e facilitam a descamação. Podem ocorrer efeitos adversos locais ligeiros, tais como prurido transitório, irritação ou sensação de picadas.
cloreto de sódio, ureia, acetato de vitamina e, glicerol e vaselina podem ser utilizados como Hidratantes e lubrificantes. Em doentes com descamação espessa e hiperqueratose marcada, podem ser adicionados 1 ou mais agentes queratolíticos, tais como ácidos α-hidroxi (ácido láctico e glicólico),130 ácido salicílico, N-acetilcisteína,131-133 ureia (>5%),134 e propilenoglicol. Também são utilizados moduladores da diferenciação queratinocitária. Estes incluem retinóides tópicos (tretinoína, adapaleno, tazaroteno),135,136 calcipotriol,137 e dexpantenol.Retinóides tópicos muitas vezes causam irritação e pequenas fissuras muito dolorosas.Além disso, existe um risco de absorção e teratogenicidade em mulheres férteis se estas forem utilizadas de forma excessiva.138 para aumentar a eficácia dos queratolíticos e dos hidratantes, pode aplicar-se tratamento oclusivo em áreas específicas refractárias ao tratamento.Um efeito aditivo ou sinérgico também pode ser obtido pela combinação de 2 ou mais agentes queratolíticos ou hidratantes.O tratamento deve ser optimizado para cada indivíduo, dada a natureza altamente variável da situação e a sensibilidade cutânea e as diferenças na resposta a cada tratamento. O processo de otimização pode ser ajudado tratando um lado do corpo de forma diferente do outro para permitir comparações. Os recém-nascidos e as crianças pequenas devem ser tratados com um veículo sem quaisquer substâncias activas, uma vez que a pele é muito fina e sensível e a maioria dos queratolíticos não são tolerados. Além disso, o risco de absorção percutânea de produtos tópicos como ureia, ácido salicílico e ácido láctico é maior.143-145
o tratamento sistémico
os retinóides orais têm efeitos queratolíticos que ajudam a eliminar as escamas e a evitar hiperqueratose excessiva. Tanto a isotretinoína como os retinóides aromáticos (acitretina e etretinato) revelaram-se eficazes no tratamento do ARCIs.128, 146, 147 acitretina numa dose de 0, 5 a 1 mg/kg/d é o fármaco mais utilizado, especialmente em doentes com LI.148 doentes com CIE podem ter uma resposta mais completa e em doses mais baixas.
os principais efeitos adversos são perturbações mucocutâneas, teratogenicidade, perturbações músculo-esqueléticas, perfil lipídico anormal e elevação das transaminases.149-152 no que diz respeito à teratogenicidade, no caso do etretinato e acitretina, os medicamentos devem ser evitados durante a gravidez e as doentes devem evitar engravidar durante 3 anos após a interrupção do tratamento.A isotretinoína tem uma semi-vida mais curta e é completamente eliminada do organismo após 1 mês, pelo que pode ser a opção preferida em mulheres que desejem engravidar.A monitorização do tratamento deve incluir um trabalho laboratorial com teste da função hepática e perfil lipídico antes de iniciar o tratamento, depois 1 mês e de 3 em 3 meses após o início do tratamento. Nas mulheres em idade fértil, deve ser realizado um teste de gravidez nas 2 semanas anteriores ao início do tratamento e deve ser utilizada uma medida contraceptiva eficaz entre as 4 semanas anteriores ao tratamento e os 3 anos seguintes (no caso da acitretina). Quando é necessário um tratamento prolongado com retinóides, o crescimento e o desenvolvimento ósseo devem ser monitorizados. Alguns autores sugerem a realização de um estudo ósseo antes do tratamento seguido de um exame anual.As orientações recentes não recomendam a realização de radiografia de rotina devido aos possíveis efeitos nocivos.Em vez disso, são recomendados estudos radiográficos selectivos em doentes com dor óssea atípica.152
uma alternativa ao tratamento sistémico do retinóide é o uso de fármacos conhecidos como bloqueadores do metabolismo do ácido retinóico, que aumentam os níveis endógenos de ácido retinóico. Um desses medicamentos é o liarozole, ao qual foi concedido o estatuto de órfão para o tratamento de LI, CIE e HI pela Agência Europeia de medicamentos e pela Food and Drug Administration dos EUA.153-155 este medicamento demonstrou ser mais eficaz do que o acitretin nos ensaios clínicos e é também melhor tolerado e tem um perfil farmacocinético melhor.154
outros Cuidados Médicos
em doentes com ectropião, a aplicação de lágrimas artificiais e lubrificantes oculares e hidratação da pele da face e das bochechas em particular pode reduzir a retracção palpebral. Correção cirúrgica é uma opção válida em casos graves, mas isso geralmente tem que ser repetido alguns anos depois. A hidroterapia pode ser benéfica.Os doentes devem ser aconselhados a evitar uma actividade física extenuante quando a temperatura ambiente for elevada, uma vez que a hipohidrose acarreta o risco de acidente vascular cerebral e convulsões. Retinóides orais podem melhorar a termorregulação.A fisioterapia é importante para prevenir a contractura de flexão, particularmente no caso do HI. A limpeza Regular do canal auditivo externo por um especialista em otorrinolaringologia pode prevenir a acumulação de escamas e assim prevenir a perda auditiva.
o Aconselhamento Genético e o Diagnóstico pré-natal
Quando um paciente é diagnosticado com ictiose, ele ou ela deve ser oferecido aconselhamento genético apropriado em que a natureza do transtorno, o modo de transmissão e o risco de futuras manifestações na família são explicados. O diagnóstico pré-natal pode indicar se o feto é afetado e, se este for o caso, preparação psicológica da família pode ser oferecido e problemas antecipados durante a gravidez e nascimento. Os pais podem ter a opção de abortar se não houver tratamento disponível. Além disso, se a terapia genética para estas condições se tornar disponível no futuro, o diagnóstico pré-natal permitiria a aplicação desta terapia O MAIS CEDO POSSÍVEL.
por mais de 20 anos, o diagnóstico pré-natal foi realizado tomando uma amostra de biópsia da pele fetal e estudando-a por microscopia óptica, microscopia eletrônica ou imunohistoquímica.158 159 este procedimento invasivo só poderia ser realizado nas fases tardias da gravidez, entre as semanas 15 e 23 de gestação, e foi associado com um risco de 1% a 3% de perda do feto.160,161 a identificação dos mecanismos moleculares das doenças hereditárias da pele permitiu um diagnóstico muito anterior baseado em técnicas genéticas.O ADN Fetal 102,162–164 é obtido por amniocentese realizada entre as semanas 15 e 20 ou por amostragem coriónica villus entre as semanas 10 e 12. O risco de perda fetal com estas técnicas é inferior a 0,5% a 1%.Outros métodos não invasivos no desenvolvimento são a análise do DNA das células fetais e o DNA fetal livre na circulação materna 166, bem como o uso de ultrassom tridimensional.167,168
o diagnóstico genético pré-implantação também pode ser possível em técnicas de fertilização in vitro, de modo que apenas ovos fertilizados livres da mutação são implantados no útero, evitando assim a necessidade de aborto na maioria dos casos.169
estratégias futuras para o tratamento genético da ictiose
embora tenham sido feitos progressos importantes no diagnóstico genético da ictiose, estão também a ser desenvolvidas novas estratégias para estas doenças.170 a pele é o órgão mais acessível para terapias de transferência de genes, e assim tais técnicas são minimamente invasivas.171 no entanto, a pele também tem características imunológicas únicas que não favorecem a expressão de longo prazo de um produto transgênico.172 In LI, a process of ex vivo gene transfer managed to restore normal TGM1 expression and correct the phenotype of skin transplanted on the back of imunossupressed mice.173.174 recentemente, o fenótipo de queratinócitos cultivados de doentes com HI devido a mutações no gene ABCA12 também foi recuperado.3
conflitos de interesses
os autores declaram que não têm conflitos de interesses.