Fronteiras em Neurologia
Introdução
Neuroacanthocytosis é um termo superordenado para um grupo de síndromes raras que são caracterizados por sintomas neurológicos em combinação com picos deformado células vermelhas do sangue (acanthocytes). Este relatório centra-se na Corea-acantocitose (CCA) , que é uma doença órfã com cerca de 1000 indivíduos afectados em todo o mundo devido a mutações autossómicas-recessivas no gene VPS13A (vacuolar protein sorting 13 homolog a) do cromossoma 9q (1). A doença tem um curso progressivo, as causas de aumento da mortalidade podem ser diminuídas funções motoras correlacionadas com condições de risco tais como disfagia, mas também mortes inesperadas súbitas foram descritas (2, 3). Até agora, não há tratamento causal.
a coexistência suspeita de acantocitose e distúrbios de movimento foi relatada pela primeira vez na década de 1970 por Levine e Critchley (4, 5) e desde então tem sido aceite como a característica proeminente da doença. No entanto, com o nosso relatório de caso, descrevemos um raro fenótipo clínico de ChAc sem distúrbios de movimento óbvios, sugerindo uma variedade mais ampla de apresentações clínicas. Ferramentas de diagnóstico têm que enfrentar esses desafios para estabelecer o diagnóstico correto, nós aqui sugerimos neuroimagem molecular como um método Chave.
Case Report
the male index patient first presented at 25 years of age with two unpoked bilateral tónico clonic peasons. Não havia histórico médico. A ressonância magnética cerebral e o EEG nessa idade eram normais e não foi iniciado tratamento devido à relutância do doente e a eventos Pouco frequentes. No entanto, o paciente tinha convulsões em curso agora apresentando-se como epilepsia parcial. Ele descreveu sentimentos recorrentes de tonturas súbitas que foram diagnosticadas como uma aura vertiginosa. Além disso, ele teve convulsões discognitivas nas quais mostrou (a) falta de resposta e recitação de orações Turcas, (B) Automatismos orais, ou (C) mexer com as mãos e proferir sons—tudo em parte evoluindo em convulsões tônicas Clônicas. O vídeo-EEG agora mostra complexos de ondas de espigões locais, tanto no lóbulo temporal direito como esquerdo. De acordo com a semiologia das convulsões, foi feito o diagnóstico de epilepsia do lobo temporal bilateral e foi iniciada a medicação com levetiracetam.
na idade de 31 crises não foram totalmente suprimidas, como parte de outros diagnósticos de epilepsia do lobo temporal, o paciente passou por um FDG-PET (Figura 1) que mostrou um hipometabolismo mesiotemporal bilateral, em linha com a origem das crises mesiotemporais. No entanto, houve também um marcado, altamente incomum hipometabolismo estriatal que levantou a suspeita de uma perturbação neurodegenerativa do movimento. Consequentemente, foi iniciada uma extensa avaliação de acompanhamento (ver Figura 2). Foi realizado um FP-CIT-SPECT que produziu uma perda moderadamente bilateral da disponibilidade do transportador estriado de dopamina, em linha com um declínio da integridade nigrostriatal (Figura 1). Uma ressonância magnética de alta resolução apresentou agora atrofia bilateral do núcleo caudatus (Figura 3). O exame neurológico mostrou um padrão discreto de marcha e hipomimia, mas nenhum outro afeto do sistema motor extrapiramidal e nenhum sintoma bulbar como a alimentação de distonia. Para além do baixo estado de reflexo nos membros inferiores, para o qual os estudos de condução nervosa confirmaram uma polineuropatia axonal sensorimotor, o exame neurológico foi normal. Os sintomas neuropsicológicos incluíram aspectos de uma perturbação de ansiedade e o doente relatou dificuldades com a perda de memória a curto prazo. Testes neuropsicológicos, no entanto, não confirmaram um mau funcionamento mnéstico manifesto, mas sim uma distração inespecífica, que pode ter sido aumentada por supressão insuficiente de convulsões na época. O teste laboratorial foi negativo para qualquer epilepsia sintomática (i.e.( anticorpos anti-encefalite auto-imune, anticorpos antineuronais). O líquido cefalorraquidiano não mostrou sinais de inflamação, mas um aumento moderado da proteína (765 mg/l). A análise de aminas biogénicas no líquido cefalorraquidiano revelou uma elevação da glutamina que associámos a crises recentes. No sangue periférico, foram encontrados 0, 4% dos acantócitos. O teste serológico da doença de Wilson deu negativo. Notável foi uma elevação persistente da creatina cinase (intervalo: 1.000–3. 000 U/l) que levou ao diagnóstico suspeito de uma doença mitocondrial. Para outras investigações, foi realizado um teste de lactato isquêmico que mostrou resultados normais. Foi realizada uma biópsia muscular do M. gastrocnemicus, mas a análise da função mitocondrial e da cadeia respiratória foi normal.
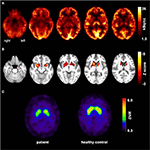
Figura 1. Resultados de estudos de imagiologia molecular. As imagens transaxiais de FDG-PET (a) mostraram não só um ligeiro hipometabolismo mesiotemporal bilateral (em linha com a origem das convulsões mesiotemporais), mas também um acentuado hipometabolismo estriatal que se verificou ser altamente significativo em comparação com controlos saudáveis . Um exame adicional FP-CIT-SPECT (C) também revelou uma perda moderada de transportadores estriados de dopamina, testemunhando um declínio da integridade nigrostriatal (um controle saudável combinado com a idade é mostrado para comparação; DVR, razão de volume de distribuição).

Figura 2. Curso clínico e de diagnóstico do paciente index.
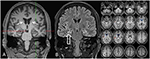
Figura 3. A ressonância mostra atrofia da cabeça caudal bilateral discreta e um hipocampo do lado direito menor e hiperintenso, indicando esclerose hipocampo do lado direito . O hipocampo esquerdo é um pouco hiperintenso, mas não atrófico. A atrofia da cabeça caudal Bilateral é confirmada com a análise CVR, na qual as cores azuis mostram cores cinzentas e vermelhas diminuídas aumento do volume do líquido cefalorraquidiano, respectivamente (C).
a história da família revelou uma consanguinidade de seus pais (primos em primeiro grau), que eles mesmos não tinham história médica significativa. Dos seus seis irmãos, dois (entre os 20 e 30 anos de idade) também já tinham sofrido ataques gerais (ver Figura 4). Os registos médicos dos seus irmãos não estavam disponíveis para nós.
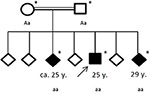
Figura 4. Pedigree da família afectada, com a idade da primeira crise epiléptica em anos (y). Seta: patente do Índice. Asterisco: testes genéticos realizados. Aa, heterozigous; aa, homozigous para C. 4326 T >A (P. Tyr1442*).Sob a hipótese de um distúrbio hereditário do movimento neurodegenerativo, o paciente e sua família foram encaminhados para testes genéticos. A sequenciação de Exome revelou uma mutação truncante homozigótica C. 4326 T >A (P. Tyr1442*) no gene ChAc VPS13A nos três irmãos afetados. Os pais consanguíneos foram ambos detectados heterozigóticos. Não foram identificadas outras variantes ou mutações no sequenciamento exome.
na verdade, em um acompanhamento com a idade de 33 anos, o paciente índice ainda tinha apenas sintomas extrapiramidais marginais e nenhum estava presente em qualquer uma de suas irmãs. Ele agora deu a impressão de ligeira rigidez apenas sob a primagem do membro superior direito, e uma leve bradicinesia dos Membros Superiores. Durante a terapêutica com levetiracetam e lacosamida, o doente não teve convulsões.
discussão
com o presente caso, demonstramos que o CCA pode se manifestar clinicamente com epilepsia do lobo temporal sem quaisquer sintomas motores significativos devido a uma nova mutação correspondente no gene VPS13A.
o curso da doença é tipicamente caracterizado por uma perturbação progressiva do movimento (incluindo Corea, distonia, parkinsonismo), alterações cognitivas e psiquiátricas e sintomas miopáticos com marcadores serológicos de acantocitose e hiperckemia. As convulsões já foram notadas como um sintoma de ChAc (6), mas os distúrbios do movimento ainda são considerados o principal sintoma. Evidencia-se que a epilepsia, e mais especificamente a epilepsia originária do lobo temporal, pode ser um fenótipo subestimado do ChAc. Peluso et al. relatou um paciente semelhante ao nosso, que, mesmo com a idade de 46 anos, não mostrou distúrbios de movimento ao apresentar resultados de neuroimagem indicativos de envolvimento de gânglios basais (7). De acordo com isso, Scheid et al. descreveu três pacientes que sofriam de epilepsia e esclerose do lobo temporal mesial (com base em estudos de ressonância magnética) como o sintoma predominante e foram diagnosticados com CAC (8). Além disso, Mente et al. recentemente forneceu os resultados da autópsia de um paciente que apresentou não só atrofia dos gânglios basais, mas também esclerose hipocampal (9). De acordo com o nosso caso, o envolvimento bilateral do hipocampo parece ser um achado comum (10, 11). A relação exata entre convulsões e esclerose é, no entanto, ainda uma questão de galinha ou ovo. O papel correlativo da coreina no desenvolvimento estrutural do hipocampo ainda não é totalmente compreendido. Curiosamente, em um modelo de mouse de mudança estrutural ChAc foi visto, como a expressão da proteína hipocampal do receptor GABA (a) gama 2 e sua coreina de proteína de ancoragem foram aumentadas (12).
na rotina clínica, este fenótipo ainda pode ser desafiador em encontrar o diagnóstico correto. A primeira dica crucial para uma doença neurodegenerativa em nosso paciente foi um declínio proeminente no metabolismo da glicose estriatal e um declínio moderado na integridade nigrostriatal em FDG-PET e FP-CIT-SPECT, respectivamente. A pequena coleção de resultados de imagiologia funcional, com base na descrição de séries de casos, mostram que as alterações no metabolismo e disfunção dopaminérgica ocorrem principalmente no núcleo caudado e putâmen (13). A ressonância magnética cerebral revelou atrofia do núcleo caudatus durante o curso da doença, que tem sido descrito como um achado típico no ChAc (14). Assim, características neurológicas distintas geralmente incluem Corea, distonia alimentar, discinésias orofaciolíngües, e tiques, juntamente com outros sintomas de afeição de gânglios basais, tais como parkinsonismo e distonia (3). É tentador especular que as alterações estriadas observadas em nosso paciente ainda estavam abaixo do limiar de causar sintomas (ou seja, achados de imagem prodromal), que podem finalmente ocorrer à medida que a doença progride. Por conseguinte, encorajamos a imagiologia estrutural e molecular como uma ferramenta de diagnóstico sensível nesta doença órfã.
no CCA, existem descrições de contagens de acantócitos entre 5 e 50% (3), mas existem alguns relatos de casos que demonstram que os acantócitos podem aparecer apenas tardiamente no curso da doença (15), podem ser muito baixos, ou mesmo estar completamente ausentes (16). Portanto, os acantócitos não devem ser considerados obrigatórios para a realização do diagnóstico. Além disso, a epilepsia pode retardar o diagnóstico, uma vez que disfarça outros sintomas típicos de caca. Em particular, os sintomas psiquiátricos, tais como alterações da personalidade e deterioração cognitiva, que são muito comuns, podem ser mal interpretados e atribuídos a efeitos secundários relacionados com o medicamento (ou seja, do levetiracetam) ou relacionados com convulsões. A hipercemia secundária é observada após convulsões tónico-clónicas gerais e uma elevação persistente pode ser negligenciada.
nosso caso também destaca o benefício crucial da sequenciação de exome para estabelecer um diagnóstico correto, especialmente em doenças raras ou quando os sintomas são vagos. A família de genes vps13 (A–D) de mamíferos tem um interesse crescente e mutações foram identificadas em outras doenças neurológicas, tais como doença de Parkinson recessiva autossômica ou ataxia espinocerebelar autossômica recessiva (17). Mutações recessivas em VPS13D causam distúrbios do movimento na infância (18). A patofisiologia subjacente do ChAc ainda não foi totalmente decifrada, mas foi mostrado que o VPS13A codifica a coreina proteica que é expressa ubiquitosamente no cérebro e em um vasto número de outros tecidos (19). Estudos revelam que participa em vias de sinalização que regulam a arquitetura do citoesqueleto, exocitose e sobrevivência celular (20). Outra mutação que foi descrita como apresentando um fenótipo dominado por convulsões em 9 doentes com CAC é a mutação C. 2343del no gene VPS13A (21). É razoável supor que mutações específicas no mesmo gene resultam em uma certa aberração da coreina que causa um fenótipo único, por exemplo, afetando diferentes áreas do cérebro. No entanto, a patofisiologia subjacente ainda é apenas especulativa e mais documentação sobre as mutações causadoras será de interesse. Mostramos aqui que a mutação truncante C. 4326 T >A( P. Tyr1442*), que não foi descrita anteriormente, resulta em um fenótipo semelhante com epilepsia predominante em todos os membros da família afetados.
observações conclusivas
a epilepsia (particularmente a epilepsia do lobo temporal bilateral) é provável que seja um fenótipo predominante do CCA. Portanto, uma busca por bandeiras vermelhas adicionais (tais como hypercemia, história familiar, polineuropatia) deve ser cuidadosamente realizada. Em casos suspeitos, a procura de acantócitos no sangue e ressonância magnética cerebral deve ser adicionada, uma vez que estas ferramentas estão amplamente disponíveis. Em caso de dúvida, os diagnósticos devem ser complementados com FDG-PET e/ou FP-CIT-SPECT, uma vez que são sensíveis na detecção do envolvimento estriado. Em epilepsias recessivas autossómicas com resultados indicativos para uma doença neurodegenerativa VPS13A teste único gene ou corein Western blot deve então ser usado para estabelecer o diagnóstico certo. Este último também deve ser considerado em outras doenças neurodegenerativas sem etiologia geneticamente definida.O consentimento informado por escrito foi obtido do paciente para a participação no estudo. Foi obtido consentimento informado por escrito do paciente para a publicação deste relatório de caso.
contribuições dos autores
JW, MR, e SK participaram na gestão de doentes. JW escreveu o primeiro rascunho do manuscrito. If, HU, and PM prepared figures. Todos os autores contribuíram para a revisão do manuscrito, leram e aprovaram a versão submetida.
Declaração de conflito de interesses
HU é accionista da Veobrain Gmbh, uma filial do Centro Médico Universitário de Freiburg.
os restantes autores declaram que a investigação foi realizada na ausência de quaisquer relações comerciais ou financeiras que possam ser interpretadas como um potencial conflito de interesses.
1. Walterfang m, Evans A, Leong Looi JC, Jung HH, Danek a, Walker RH, et al. A Neuropsiquiatria das síndromes de neuroacantocitose. Neurosci Biobehav Rev. (2011) 35:1275-83. doi: 10.1016 / j. neubiorev.2011.01.001
PubMed Abstract / CrossRef Full Text | Google Scholar
2. Walker RH. Gestão de síndromes de neuroacantocitose. Tremor Outra Hiperquinete. Movimento. (2015) 5:346. doi: 10.7916/D8W66K48
PubMed Abstract / CrossRef Full Text | Google Scholar
3. Velayos Baeza a, Dobson-Stone C, Rampoldi L, Bader B, Walker RH, Danek A, et al. Corea-Acantocitose. Generevews®. Seattle, WA: University of Washington (1993).
4. Critchley EM, Clark DB, Wikler A. Acantocitose e distúrbio neurológico sem Betalipoproteinemia. Arch Neurol. (1968) 18:134–40.Resumo Publicado
5. Levine IM, Estes JW, Looney JM. Doença neurológica hereditária com acantocitose. Uma nova síndrome. Arch Neurol. (1968) 19:403–9.
PubMed Abstract | Google Scholar
6. Hardie RJ, Pullon HW, Harding AE, Owen js, Pires M, Daniels GL, et al. Neuroacantocitose. um estudo clínico, hematológico e patológico de 19 casos. Brain(1991) 114 (Pt 1A):13-49.
PubMed Abstract | Google Scholar
7. Peluso S, Bilo L, Esposito M, Antenora a, Rosa ADR, Pappata S, et al. Corea-acantocitose sem Corea: expansão do fenótipo clínico. Parkinson Relat Disord. (2017) 41:124–6. doi: 10.2016 / J. parkreldis.2017.05.013
PubMed Abstract | CrossRef Full Text | Google Scholar
8. Scheid R, Bader B, Ott DV, Merkenschlager a, Danek A. desenvolvimento de epilepsia do lobo temporal mesial em Corea-acantocitose. Neurology (2009) 73:1419-22. doi: 10.1212 / WNL.0b013e3181bd80d4
PubMed Abstract | CrossRef Full Text | Google Scholar
9. Mente K, Kim SA, Grunseich C, Hefti MM, Crary JF, Danek A, et al. Esclerose hipocampal e epilepsia do lobo temporal mesial em Corea-acantocitose: um caso com avaliação clínica, patológica e genética. Neuropathol Appl Neurobiol. (2017) 43:542–6. doi: 10.1111 / nan.12403
PubMed Abstract / CrossRef Full Text | Google Scholar
10. Bader B, Vollmar C, Ackl N, Ebert a, la Fougère C, noachtar S, et al. Epilepsia do lobo temporal Bilateral confirmada com EEG intracraniano em Corea-acantocitose. Apreensão (2011) 20:340-2. doi: 10.1016 / j. convulsão.2010.12.007
PubMed Abstract | CrossRef Full Text | Google Scholar
11. Marson AM, Bucciantini E, Gentile e, Geda C. Neuroacantocitose: resultados clínicos, radiológicos e neurofisiológicos numa família italiana. Neurol Sci. (2003) 24:188–9. doi: 10.1007 / s10072-003-0123-1
PubMed Abstract | CrossRef texto completo | Google Scholar
12. Kurano Y, Nakamura M, Ichiba M, Matsuda m, Mizuno e, Kato M, et al. A deficiência da coreina leva à regulação do receptor GEFIRINA e GABA(A). Biochem Biophys Res Commun. (2006) 351:438–42. doi: 10.1016 / j. bbrc.2006.10.070
PubMed Abstract | CrossRef Full Text | Google Scholar
13. Ehrlich DJ, Walker RH. Functional neuroimaging and chorea: a systematic review. J Clin Mov Disord. (2017) 4:8. doi: 10.1186 / s40734-017-0056-0
PubMed Abstract | CrossRef texto completo | Google Scholar
14. Connolly BS, Hazrati L-N, Lang AE. Resultados neuropatológicos em Corea-acantocitose: novos conhecimentos sobre os mecanismos subjacentes ao parkinsonismo e às convulsões. Acta Neuropathol. (2014) 127:613–5. doi: 10.1007 / s00401-013-1241-3
PubMed Abstract | CrossRef texto completo | Google Scholar
15. Sorrentino G, de Renzo a, Miniello S, Nori o, bonita V. aparecimento tardio de acantócitos durante a corea-acantocitose. J Neurol Sci. (1999) 163:175–8.
PubMed Abstract | Google Scholar
16. Bayreuther C, Borg M, Ferrero-Vacher C, Chaussenot A, Lebrun C. Coréo-acantocitose sem acantócitos. Revue Neurol. (2010) 166:100–3. doi: 10.1016 / j. neurol.2009.03.005
CrossRef Full Text | Google Scholar
17. Lesage S, Drouet V, Majounie e, Deramecourt V, Jacoupy M, Nicolas a, et al. A perda da função VPS13C no parkinsonismo autossômico-recessivo causa disfunção mitocondrial e aumenta a mitofagia dependente de PINK1/Parkin. Am J Hum Genet. (2016) 98:500–13. doi: 10.1016 / j. ajhg.2016. 01. 014
PubMed Abstract | CrossRef Full Text | Google Scholar
18. Gauthier J, Meijer IA, Lessel D, Mencacci NE, Krainc D, Hempel M, et al. Mutações recessivas em >VPS13D causam distúrbios de movimentos na infância. Ann Neurol. (2018) 83:I6. D. O. I.: 10.1002 / ana.25204
PubMed Abstract / CrossRef Full Text | Google Scholar
19. Kurano Y, Nakamura M, Ichiba M, Matsuda m, Mizuno e, Kato M, et al. Distribuição In vivo e localização da corein. Biochem Biophys Res Commun. (2007) 353:431–5. doi: 10.1016 / j. bbrc.2006.12.059
PubMed Abstract | CrossRef Full Text | Google Scholar
20. Lang F, Pelzl L, Schöls L, Hermann a, Föller M, Schäffer TE, et al. Neurônios, eritrócitos e além-as diversas funções da coreina. Neurosignals (2017) 25:117-26. doi: 10.1159/000485457
PubMed Abstract | CrossRef texto completo | Google Scholar
21. Benninger F, Afawi Z, Korczyn AD, Oliver KL, Pendziwiat M, Nakamura M, et al. Convulsões como sintoma apresentando e proeminente na Corea-acantocitose com mutação genética C. 2343del VPS13A. Epilepsia (2016) 57:549-56. doi: 10.1111/epi.13318
PubMed Abstract | CrossRef Full Text | Google Scholar