Frontiers in Pediatrics
- Introduction
- síndrome de QT longo
- LQTS1
- LQTS2
- LQTS3
- LQTS4
- LQTS5
- LQTS6
- LQTS7
- LQTS8
- LQTS9 e LQTS10
- LQTS11
- LQTS12
- LQTS13
- Adquirida LQTS
- Eletrocardiográfica Recursos nos Três Formas Comuns de Síndrome do QT Longo
- genotipo-fenótipo
- Gestão Clínica de LQTS
- LQTS: Perspectiva Saudita
- Declaração de conflito de interesses
- agradecimentos
Introduction
family or hereditary cardiac arritmias compreendem percentagens significativas de arritmias e também causais a morte cardíaca súbita (SCD) (1, 2). Durante as duas últimas décadas, cientistas e clínicos têm dado um enorme esforço para desvendar os mecanismos complexos e intrincados das arritmias familiares congênitas (1-8). Para compreender o mecanismo da arritmogénese, precisamos de saber os fundamentos da estrutura celular Cardíaca e as suas propriedades electrofisiológicas. Os miócitos cardíacos são as principais células funcionais do coração e estão extensivamente acoplados de modo que os impulsos se propagam rápida e uniformemente. Os cardiomiócitos são separados uns dos outros por um limite especializado chamado disco intercalado; proteínas de junção gap, desmossomas cardíacos e canais iônicos estão localizados no disco intercalado (9). As junções de Gap consistem em conectinas apertadas que permitem a troca intercelular de pequenas moléculas e também permitem fluir as correntes excitatórias de uma célula para a sua célula vizinha. Os desmossomas, juntamente com as junções de adesão, são responsáveis pela fixação mecânica dos cardiomiócitos individuais. Todos estes componentes nos discos intercalados são sequencialmente segregados e cada componente exerce a sua função única; a ruptura de um componente afecta a função de outros componentes, o que predispõe o coração a desenvolver arritmias (9-11). Os canais de iões cardíacos são complexos proteicos que formam poros que fornecem voltagem fechada e intrinsecamente coordenada para dentro e para fora do movimento das correntes iônicas através das membranas celulares, essencial para a geração e propagação do ritmo cardíaco. Síndrome do QT longo (LQTS), síndrome do QT curto (SQTS), síndrome do seio doente (SSS), condução cardíaca defeito (CCD), BrS, catecholaminergic taquicardia ventricular polimórfica (CPVT), repolarização precoce, síndrome de (ERS), e familiares de Fibrilação Atrial (FA) são conhecidos atualmente cardíaca channelopathies, o que pode ocorrer devido a um ou a vários defeitos nos genes ligados ao ritmo cardíaco geração e propagação.
nesta revisão, iremos descrever exclusivamente sobre arritmias relacionadas com LQTS, a sua fisiopatologia e gestão clínica actualmente disponível. Temos sido pioneiros na elucidação da patologia genética em arritmias cardíacas familiares na Arábia Saudita (1, 12-14), ainda estamos realizando investigações cardiogenéticas na Arábia Saudita, no final desta revisão, vamos discutir o conhecimento atualmente disponível sobre os achados genéticos e clínicos obtidos de várias famílias da Arábia Saudita com história de síncope e SCD. Acrescentaremos também os dados recentemente publicados sobre LQTS de uma equipa do King Faisal Specialist Hospital and Research Centre, Riyadh (15).
síndrome de QT longo
LQTS congénitos é uma doença hereditária definida pelo prolongamento do intervalo QT no electrocardiograma (ECG). Os doentes com todas as formas de LQTS estão predispostos à taquiarritmia ventricular, torsades de pointes (TdP) conduzindo a síncope recorrente ou DCOPE. Em muitos casos, síncope ou morte súbita pode ser a primeira e a única manifestação. O LQTS afeta cerca de 1 em cada 2.000 pessoas em todo o mundo (16). A característica distintiva do LQTS é o prolongamento do intervalo QT no ECG (corrigido para a frequência cardíaca, isto é, QTc). Os valores normais de QTc são 440 ms nos machos e 450 ms nas fêmeas. Nas crianças, os valores dependentes da idade e do sexo são relevantes. Recomendações de consenso recentes para o diagnóstico de LQTS são as seguintes:(17, 18):
1. LQTS é diagnosticado:
um. Na presença de um LQTS classificação de risco ≥3.5 na ausência de uma causa secundária para o prolongamento QT e/ou
b. Na presença de um inequivocamente patogênicos mutação em um dos LQTS genes ou
c. Na presença de um intervalo QT corrigido para a frequência cardíaca utilizando a fórmula de Bazett (QTc) ≥500 ms em electrocardiograma de 12 condutores repetidos e na ausência de uma causa secundária para o prolongamento do intervalo QT.
2. Os LQTS podem ser diagnosticados na presença de um QTc entre 480 e 499 ms em ECGs de 12 chumbo repetidos num doente com síncope inexplicável na ausência de uma causa secundária para o prolongamento do intervalo QT e na ausência de uma mutação patogénica.
os doentes com duração do intervalo QTc ≥500 ms tiveram uma probabilidade cumulativa significativamente superior de apresentarem a sua primeira síncope em comparação com os doentes com duração do intervalo QTc <500 ms desde o nascimento até aos 20 anos de idade (19). Mas, os doentes que tiveram episódios de 2, 3 e 4 síncope, o risco de um episódio subsequente de síncope foi virtualmente idêntico entre os doentes que tinham uma duração de QTc estreita ou prolongada (19). A base molecular de LQTS é heterogênea e até o momento, mutações em 13 genes diferentes foram descritas causais a LQTS (1-8, 19, 20). Um defeito genético é geralmente encontrado em 70% dos pacientes LQTS em um desses 13 genes (1-8, 19, 20). Entre os 13 tipos de LQTS atualmente conhecidos, os mais comuns são LQTS1, LQTS2, e LQTS3, devido a defeitos nos genes do canal cardíaco, KCNQ1, KCNH2, e SCN5A, respectivamente. Noventa por cento de todas as mutações causais LQTS são encontradas nestes três genes (1-8, 19, 20). As mutações nos restantes 10 genes são raras e compreendem apenas ∼10% de todas as mutações LQTS actualmente conhecidas.
síndrome de QT longo é geralmente uma doença autossômica dominante, mas, ocasionalmente, mutações múltiplas em um único gene ou em genes diferentes podem ser encontradas em 5-10% dos pacientes com LQTS (21, 22). Pacientes com múltiplas mutações poderiam apresentar um tempo de QTc em comparação com aqueles com uma única mutação e pacientes também estão na ∼3.5 vezes maior risco de vida risco de eventos cardíacos (21, 22)
LQTS1
Mutações no gene KCNQ1 são as mais comuns de todas LQTS e referido como LQTS tipo 1 (LQTS1). 50% de todas as mutações causais LQTS são encontradas no gene KCNQ1 (20). KvLQT1 (também chamado de Kv7.1) é uma proteína feita pelo gene KCNQ1 e as formas de proteína tetramers no retículo endoplasmático no interior das células, que supostamente co-monta com a proteína de vison (codificado por KCNE1) e, em seguida, são transportadas para a membrana plasmática dos miócitos cardíacos, onde eles mediar um lentamente de ativação atual, o que acelera a repolarização do potencial de ação cardíaco tecidos, esta corrente é conhecida como IKs (Figura 1). Lqts1 mutações causais KCNQ1 são na sua maioria mutações sem sentido e, em casos raros, podem ser mutações de transição na região C-terminal (23). Arritmia cardíaca nos portadores de mutação KCNQ1 são desencadeados por impulsos adrenérgicos, por exemplo, estresse emocional, esforço físico, mergulho, natação (24-26). Os doentes com mutações nos domínios transmembranares do KvLQT1 apresentam um risco mais elevado de acontecimentos cardíacos relacionados com o LQTS e têm uma maior sensibilidade à estimulação simpática (26, 27). Os doentes com mutações sem sentido apresentam um risco aumentado em comparação com os doentes com mutações sem sentido ou truncantes (27, 28). A variação no gene 3′ – UTR do kcnq1 também afeta significativamente a suscetibilidade à arritmia, presumivelmente por impacto na expressão do gene (29). Doentes com arritmias devidas a mutações KCNQ1 respondem muito bem aos β-bloqueadores, mas alguns doentes ainda podem ser menos sensíveis ou mesmo resistentes a este medicamento. Num artigo recente, Barsheshet et al. (30) alegou que os doentes com mutações fora da região do ciclo citoplásmico (Ciclo-c) na KvLQT1 são menos sensíveis aos bloqueadores-β.
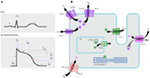
Figura 1. Correntes iónicas que contribuem para o potencial de Acção ventricular (A) e representação esquemática de um cardiomiócito que exibe (apenas) as proteínas envolvidas na patogénese de síndromes de arritmia hereditária (B). Na alínea a), o potencial de acção está alinhado com o seu tempo aproximado de acção durante o ECG. Na alínea b), ankirina-B, uma proteína adaptadora envolvida na síndrome de QT longo tipo 4, não é representada.
mutações homozigóticas ou heterozigóticas compostas no gene KCNQ1, que podem causar a forma recessiva da doença, a síndrome de Jervell e a síndrome de Lange-Nielsen (JLNS), tipo 1 (31), são bastante raras. Os doentes com NCJ sofrem também de arritmias cardíacas graves e surdez (31, 32). Doentes com mutações KCNQ1 causais a JLNS geralmente não têm quaisquer IKs funcionais (28, 31-33). É possível que alguns pacientes não tenham surdez apesar de terem mutações homozigóticas ou heterozigóticas compostas em KCNQ1, tais casos são referidos como lqts1 autossômico recessivo (13, 32). Nestes pacientes, uma pequena quantidade de corrente funcional de IKs (<10% do total de IKs) ainda pode estar presente, o que mantém a função auditiva, mas seus defeitos de ritmo cardíaco são igualmente graves como em pacientes com JLNS (13, 32).
LQTS2
este tipo de LQT é igualmente prevalente como LQTS1, representando 35-40% dos doentes com LQT com mutação detectável(1, 20, 24, 25). O KCNH2 codifica a proteína HERG (Kv11. 1), que é a subunidade α Do Retificador K+ actual (IKr), que se activa rapidamente. Mutações patogénicas neste gene que reduz a função do canal Kv11. 1 prolongam a duração do intervalo QT (Figura 1), e são causais ao LQTS2. Vinte e nove por cento dos ataques sincopais em LQTS2 ocorrem durante o repouso/sono e apenas 13% dos ataques sincopais foram relatados para ocorrer durante o exercício (25, 34). Ruídos súbitos surpreendentes, por exemplo, anéis de despertador, Campainhas, anéis de telefone, normalmente desencadeiam ataques sincopais nestes doentes (24, 34). Os doentes com mutações na região formadora de poros da Kv11.1 (codificada pelo gene KCNH2) são susceptíveis a um risco elevado de acontecimentos cardíacos relacionados com arritmia em comparação com doentes com mutações não relacionadas com poros (35). Nos recém-nascidos, o bloco Atrioventricular (AV) 2:1 está preferencialmente associado a mutações KCNH2 (36). Foram também encontrados blocos AV completos complicados por LQTS em 17% dos doentes adultos com mutação no gene KCNH2 (37). As mutações homozigóticas na KCNH2 são raras e, quando presentes, os doentes sofrem de uma forma grave de LQTS, com 2:1 bloqueio AV e arritmias ventriculares graves, durante os estágios intra-uterinos ,bem como após o nascimento (11, 38-40). Além das numerosas mutações descritas na patologia LQTS2, variantes polimórficas comuns no gene KCNH2 também podem modular a gravidade da doença. Um exemplo intrigante é o polimorfismo K897T (SNP) em KCNH2, que está presente em 33% da população em geral (41). Foi relatado que o K897T exacerbou a patogenicidade das mutações KCNH2 (42, 43).
LQTS3
Nav1.5 é a subunidade α-formadora de poros do canal cardíaco na+ dependente de voltagem, é uma proteína integral de membrana codificada pelo gene SCN5A e está envolvida no início e condução dos potenciais de ação cardíaca. Os canais cardíacos na+ são compostos por uma subunidade α-formadora de poros (codificada por SCN5A) e uma ou mais subunidades β auxiliares.
LQTS3 é causado pelo ganho de mutações de função que interrompem a inactivação rápida da subunidade α (Figura 1) e uma mutação no gene SCN5A foi descrita em <10% de todos os doentes com LQTS com mutação (1, 20, 24). Os doentes com LQTS3 experimentam a maioria (39%) dos seus acontecimentos cardíacos durante o sono/repouso (25), e cerca de 13% dos acontecimentos foram notificados durante o exercício (25). In several instances, a single SCN5A mutation was shown to exercise two or even three distinct phenotypes of arritmias in the same family, e.g., LQTS, BrS, or CCD (44-47). Os doentes do sexo masculino com mutação LQTS3 podem desenvolver sintomas muito mais cedo do que os doentes do sexo feminino (48). As mutações scl5a heterozigóticas e homozigóticas foram descritas em LQTS3 com bloco funcional 2:1 AV (49). Recentemente encontramos uma família iraniana com a mutação 1507_1509delQKP em vários membros da família, onde os pacientes tinham combinado LQTS e CCD (dados não mostrados). Esta mutação também foi notificada em doentes de outros países, o que sugere que esta é uma mutação recorrente e de ponto quente . Foram também notificadas mutações com tais características de perda e ganho de função durante diferentes fases do potencial de acção em 1493delK e em 1795 mutações de NSD (44, 51).Ocasionalmente, um SNP também pode exercer efeito patogênico em seus portadores. S1103Y é uma variante comum do gene SCN5A, presente em 13% dos afro-americanos (52). Os portadores com esta variante apresentam um risco aumentado de arritmias e síndrome de morte súbita infantil (SIDS) (52).
LQTS4
LQTS4 representa a primeira forma não-canal de LQTS. Uma mutação no ANK2, uma proteína adaptadora, leva a sobrecarga intracelular de cálcio que contribui para o LQTS4 (53, 54). Para além do prolongamento do intervalo QT, os doentes com esta síndrome podem ter bradicardia sinusal, AF paroxística e CPVT (54). O efeito patogénico das mutações ANK2 pode ser moderado a grave e as expressões clínicas dependem da gravidade da mutação.
LQTS5
as mutações em KCNE1 estão associadas com o LQTS5 (Figura 1) (55, 56). As mutações heterozigóticas do KCNE1 reduzem as IKs, exercendo um efeito negativo dominante no Alelo normal que as acompanha, e levam a uma repolarização cardíaca retardada (Figura 1), responsável pelo aumento do risco de arritmias (6). Doentes com mutações homozigóticas no KCNE1 sofrem de NJJ (tipo 2) (57, 58).
D85N é um polimorfismo no gene KCNE1, presente em 0.7-1% da população em geral (41). In a study by Nishio et al. (59) o polimorfismo de D85N foi mais frequentemente encontrado nos doentes com LQTS, tornando-se um genótipo de risco na patologia de LQTS (potencialmente apenas na população asiática). Na Europa, o D85N foi notificado em 5% dos doentes com LQTS (aLQTS) adquiridos (numa coorte de 32 doentes), que tinham experimentado TdP (60).
LQTS6
o gene KCNE2 codifica o peptídeo 1 (MiRP1) relacionado com o vison, uma subunidade beta putativa do canal cardíaco de potássio IKr (Figura 1). Mutações no gene KCNE2 podem também conduzir a defeitos no componente de ativação rápida da corrente retificadora de potássio (IKr) retardada, base patológica de LQTS6 (61). Estímulos auditivos / acústicos como ruído de despertador, campainha, etc. pode provocar ataques sincopais em portadores de mutação KCNE2, semelhantes à mutação KCNH2 (62).
LQTS7
esta síndrome é também conhecida como síndrome de Andersen–Tawil (ATS). A ATS é uma doença rara, manifestada por síncope ocasional e paragem cardíaca. Características do ECG incluem prolongamento ligeiro do intervalo QT, ondas U anormais, ectopia ventricular frequente, taquicardia ventricular bidirecional (VT) e VT polimórfica. Esta síndrome também apresenta características extracardiacais, por exemplo, paralisia periódica do músculo esquelético e problemas de desenvolvimento, tais como fenda palatina, orelhas baixas, estatura curta e defeitos de desenvolvimento nos membros (63). A maioria dos doentes com TSA clinicamente diagnosticados teve uma mutação no KCNJ2 (63). O KCNJ2 codifica uma subunidade de formação de poros que retifica internamente os canais de potássio (IK1) (Figura 1) (64, 65).
LQTS8
Também conhecida como síndrome de Timothy (TS), os pacientes mostram graves prolongation em seus ECGs, que é combinado com sindactilia, calvície no nascimento, e os dentes pequenos em 100% dos casos e menos penetrante cardíaca estrutural, malformações, retardo mental, autismo, e dismorfismo facial recursos (66). Existem dois subtipos: TS1 (clássico) e TS2 (forma rara).
TS2 é cardiologicamente mais severo do que TS1 (66, 67). Os doentes do TS2 também não têm sindactilia (67). As mutações na subunidade α – 1 da Corrente de cálcio do tipo L (ICa-L) que codifica o gene CACNA1C conduzem a ambas as formas de TS (LQTS8). No coração e no cérebro, onde o CACNA1C é predominantemente expresso, o exon-8 é encontrado em ∼80% das transcrições de mRNA e o exon-8A é presente ∼20% transcrições (66). A mutação G406R em exon-8A é causal à forma clássica de TS (TS1) e G402S em exon-8 foi notificada em severer em TS2. Uma nova adição a esta lista é a mutação Ala1473Gly, que foi descrita em um lactente com fenótipo expandido (68). Todas as mutações foram de novo ou mosaicas no progenitor, e todo o ganho resultante da função do canal ICa-L (66-69).
LQTS9 e LQTS10
mutações em CAV3 ou SCN4B produzem ganho de função no final da INa, causando um fenótipo tipo LQTS3 (70-74). São conhecidos como LQTS9 (associados à mutação CAV3) e LQTS10 (associados à mutação SCN4B).Caveolae são bem descritas por Engelman et al. (72) como “pequenas cavernas” na membrana plasmática. São pequenas fossas não revestidas e são consideradas como o local de importantes eventos dinâmicos e Regulatórios na membrana plasmática (72, 73). As caveolinas são as principais proteínas das caveolae, e a caveolin-3 (codificada pelo gene CAV3) é especificamente encontrada em cardiomiócitos e células do músculo esquelético. Vários canais iônicos cardíacos foram especificamente relatados para serem localizados nas caveolae extraídas de miócitos cardíacos que são enriquecidos em caveolin-3 (72, 73). Adicionalmente, componentes da cascata sinalizadora dos receptores β-adrenérgicos também estão presentes em membranas enriquecidas com caveolae (72, 73).
SCN4B codes for NaVß4, which is an auxiliary β-subunit of the cardiac sodium channel. Até agora, apenas uma mutação (L179F) foi relatada neste gene em uma família mexicana com múltiplos membros afetados (74). A mutação neste gene resultou num ganho de função da Corrente Nav1.5 (74).
LQTS11
no coração, a regulação simpática da duração do potencial de Acção cardíaca (APD) é mediada pela activação do receptor β-adrenérgico (β-AR), que requer a montagem de AKAP9 (Yotiao) com a subunidade α (KvLQT1) do canal IKs. Mutação no AKAP9 causa LQTS11 (75). Até à data, apenas foi notificada uma mutação, S1570L, em AKAP9 (75).
LQTS12
mutação no gene da α-1-Sintrofina (SNTN1) são causais ao lqts12. A mutação neste gene leva ao ganho de função do canal cardíaco de sódio (Nav1.5), que é a base patológica do LQTS12 (76).
LQTS13
G subunidade do canal de potássio interiormente retificante (Kir3. 4) é codificada pelo gene KCNJ5. Uma mutação de perda de função neste gene pode causar LQTS13 (77). Até agora, apenas uma mutação, G387R, foi descrita em uma família chinesa com nove pacientes tendo esta mutação. A redução da expressão da membrana plasmática de Kir3.4 foi sugerida como a patologia do LQTS nos doentes.
Adquirida LQTS
além do congênita LQTS, outra variante de LQTS conhecido como aLQTS também existe, que é causado por fatores e substâncias que diminuem o fluxo de potássio e prejudicar a capacidade de o miocárdio a repolarize. As condições bem reconhecidas são Sexo Feminino, hipocaliemia e medicamentos que inibem os canais cardíacos de potássio (78-80). Uma série de medicamentos comumente prescritos também podem se ligar preferencialmente e bloquear o canal HERG (Kv11.1, uma proteína codificada pelo gene KCNH2) e predispõe a aLQTS (79, 80). Recentemente, foi demonstrado que o bloqueio aos IKs também poderia contribuir para aLQTS induzidos por drogas, especialmente quando a reserva de repolarização é comprometida (81). Verificou-se que a fluoxetina e a norfluoxetina suprimem as propriedades da IKs, tanto in vivo como in vitro, conduzindo a LQTS marcados (81). Polimorfismos, D85N em vison (gene KCNE1), T8A, Q9E em MiRP1 (gene KCNE2), que são putativo β-sub-unidades do IKs e IKr canais, foram relatados para fazer aLQTS (82, 83). Foram também notificados LQTS auto-imunes num doente com anticorpos anti-HERG contendo IgG (84).
Eletrocardiográfica Recursos nos Três Formas Comuns de Síndrome do QT Longo
Típica de ST-T-padrões de onda estão presentes na maioria dos genotipada LQTS pacientes e pode ser usado para identificar LQTS1, LQTS2, e, possivelmente, LQTS3 genótipos (85, 86).
a forma LQTS1 do LQTS está associada a uma onda T larga sem diminuir o intervalo QT no exercício (figura 2A). O LQTS2 está associado à baixa amplitude, muitas vezes bifid, ondas T (figura 2B). O LQTS3 está associado a um longo segmento iso-eléctrico e a uma onda T estreita e alta (figura 2C). A dependência da pausa do início do TdP em LQTS congénitos é específica ao genótipo, sendo predominante em LQTS2 mas quase ausente em LQTS1 (87).

Figura 2. Gravações electrocardiográficas de pacientes com LQTS1, LQTS2 e LQTS3. A) ECG de 12 chumbo de um macho de 18 anos com uma mutação KCNQ1. O intervalo QT é prolongado (QTc = ±500 ms). O segmento ST tem uma ampla base e amplitude relativamente grande. O intervalo de condução é normal (calibração padrão). B) ECG de 12 chumbo de uma rapariga de 14 anos com uma mutação KCNH2. O intervalo QT é prolongado (QTc ± 520 ms). O segmento ST é entalado em chumbo V3 e tem uma amplitude relativamente baixa nos terminais da extremidade. O intervalo de condução é normal (calibração padrão). C) ECG de 12 chumbo de um rapaz de 12 anos com uma mutação SCN5A. O intervalo QT é prolongado (QTc ± 600 ms). O segmento ST tem um longo (quase) segmento iso-elétrico com uma onda T Grande, afiada e estreita. O intervalo de condução é normal (calibração padrão).
embora os padrões possam sugerir um genótipo específico dos LQT, foram frequentemente encontradas excepções.
genotipo-fenótipo
a síndrome de QT longo é uma doença autossómica dominante. A análise do genótipo e fenótipo entre os portadores de mutação heterozigótica tem sido conduzida bastante extensivamente nos pacientes LQTS1, LQTS2 e LQTS3. Durante a infância, o risco de acontecimentos cardíacos é significativamente maior nos indivíduos do sexo masculino LQTS1 do que nos indivíduos do sexo feminino, enquanto que não foram observadas diferenças significativas no risco de acontecimentos cardíacos relacionadas com o sexo entre os doentes com LQTS2 e LQTS3 (88-91). Durante a idade adulta (também após os 40 anos), as fêmeas LQTS1 e LQTS2 podem ter um risco significativamente maior de eventos cardíacos do que os respectivos machos (88-91). Em geral, a letalidade dos acontecimentos cardíacos parece ser predominante nos doentes com LQTS3 do que nos doentes com LQTS1 e LQTS2 (91). As mulheres com LQTS apresentam um risco reduzido de eventos cardíacos durante a gravidez, mas o risco aumenta bastante durante o período pós-parto de 9 meses, especialmente nas mulheres com mutação no gene KCNH2 (92).
morte cardíaca súbita em crianças também pode ser causada por mutações nos genes do canal iónico cardíaco (93-96). Cerca de 28% das crianças com uma DCL inexplicável (entre 1 e 18 anos, idade média: 12, 3 ± 3, 8 anos) foram Portadores de mutações nos genes causais do LQTS (97). Em SIDS, mutações em SCN5A pareciam predominantes (98, 99), mas, mutações em KCNQ1, KCNH2, KCNE2, e CAV3, SCN4B, e SCN3B também foram encontradas (100, 101). Também foram notificadas mortes intra-uterinas fetais devido a defeitos nos genes dos canais iónicos cardíacos (12, 102).
Gestão Clínica de LQTS
Cessação de todos os medicamentos que são conhecidos por prolongar o intervalo QT e também a correção do desequilíbrio eletrolítico e/ou precipitantes as condições metabólicas deve ser o foco principal durante o tratamento (adquirida) LQTS pacientes. Os sintomas no LQTS são frequentemente mediados supra-renal, pelo que a restrição da participação dos doentes em actividades atléticas é geralmente recomendada (24, 25). O principal esteio da terapia clínica para os LQTS é O β-bloqueio. As preparações de longa duração de propranololol, nadolol e metoprolol são geralmente utilizadas ,e a sua eficácia No β-bloqueio é avaliada através da diminuição da frequência cardíaca do exercício (por exemplo, através de>20%) (24, 25). Entre todos OS β-bloqueadores, o propranolol e o nadolol são considerados superiores ao metoprolol em doentes sintomáticos (103). Além disso, O β-blockade também pode ser utilizado como tratamento profiláctico em portadores de mutação silenciosa para reduzir a DSC (24). In a study by Barsheshet et al. (30), os doentes com mutações de Ciclo-C no gene KCNQ1 apresentaram um risco elevado de acontecimentos cardíacos com risco de vida e tiveram benefícios significativos do tratamento com bloqueadores-β. Uma vez que as mulheres com LQTS2 apresentam um risco aumentado durante o período pós-parto de 9 meses, os bloqueadores beta devem ser prescritos para reduzir quaisquer acontecimentos cardíacos durante este período de alto risco (92). Pode ser considerado um cardioverter–desfibrilhadores implantáveis (ICDs) em doentes com síncope recorrente apesar da terapêutica com bloqueadores-β ou em doentes com risco elevado de paragem cardíaca (p. ex., LQTS2 sintomático e LQTS3 com prolongamento documentado do intervalo QTc). Recomenda-se denervação por compaixão (LCSD) para doentes com QTL de alto risco em que um DIC é contra-indicado ou recusado e os bloqueadores-β ou não são eficazes, não tolerados, não aceites ou contra-indicados (18, 104). Os doentes com JLNS têm geralmente um QTc > 500 ms e também apresentam um risco elevado, OS β-bloqueadores têm eficácia insuficiente nos doentes em que foi recomendada uma terapêutica precoce com DCI (18, 31).
LQTS: Perspectiva Saudita
o primeiro relatório sobre os LQTS da Arábia Saudita foi publicado em 1993 a partir do Hospital das Forças Armadas de Riade (105). Quatro lactentes e crianças jovens entre 6 e 48 meses com história de convulsões recorrentes, de uma única família, foram diagnosticados como LQTS (105). A história da família revelou que dois outros membros estendidos da família tiveram episódios semelhantes de perda súbita de consciência e três membros da família morreram subitamente (105). Em todos os casos, o diagnóstico inicial foi epilepsia (105). Vários anos mais tarde, dois casos esporádicos relatados com variante comparativamente mais severa de LQTS neonatais combinados com 2: 1 bloco AV foram relatados (106, 107). Todos estes relatórios clínicos publicados não apresentavam quaisquer achados genéticos que pudessem explicar a fisiopatologia dos LQTS neles (105-107).
nós, pela primeira vez, temos relatado defeitos genéticos como uma base patológica de LQTS em pacientes similares da Arábia Saudita. Investigamos seis famílias Sauditas com história de síncope e mortes inexplicáveis súbitas de feto, recém-nascidos e crianças (12-14). O LQTS1 recessivo autossómal foi diagnosticado em crianças de duas famílias (Figura 3). O LQTS2 autossómal recessivo foi diagnosticado em duas famílias (Figura 4). Em uma família, uma paciente do sexo feminino foi diagnosticada com LQTS2 autossômico dominante (Figura 5), a paciente teve ataques sincopais durante o período de recuperação pós-parto no hospital, o que é muito comum em mulheres portadoras de mutação KCNH2. Em todos os nossos doentes, a identificação de mutações patogénicas nos genes causais dos canais iónicos cardíacos LQTS conduziu a um diagnóstico clínico confirmado, que foi mal diagnosticado como convulsões epilépticas Antes de nos terem sido referidos (13, 14) recentemente, Shinwari et al. (15) do King Faisal Specialist Hospital and Research Centre reported a LQTS1 causal KCNQ1 mutation, H258P, in a large family with 12 affected individuals. Apenas dois portadores foram sintomáticos, o seu QTc foi > 500 ms, e OS β-bloqueadores suprimiram os sintomas clínicos num doente e o segundo doente sintomático necessitou de uma DCI.
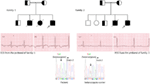
Figura 3. Desenho Genealógico da família-1 e 2 com lqts1 recessivo autossômico, probands são mostrados com uma seta. ECGs dos probands em duas famílias são mostrados no meio, com um QTc de 557 e 529 ms, respectivamente. Mutação intrónica, C. 387-5 T > a no gene KCNQ1 foi encontrada nos doentes (mostrado na parte inferior). A mutação foi mostrada com uma seta. Círculos e Quadrados não preenchidos não são portadores da mutação. Os indivíduos afetados são mostrados como círculos preenchidos (feminino) e Quadrados (masculino). Half filled squares and circles are individuals with heterozygote mutation. Os indivíduos falecidos são indicados por cortes, as varandas são indicadas por uma seta e o casamento consanguíneo é indicado por = fronteira Exon − intron é mostrado por uma linha pontilhada com seta apontando para exon.
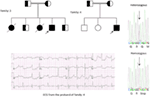
Figura 4. Desenho Genealógico da família-3 e 4 com LQTS recessivo autosómal2. O ECG do proband da família-4 é mostrado abaixo do desenho genealógico, que mostra taquicardia sinusal, bloqueio AV quase completo, grande ritmo de escape complexo com intervalos QTc muito longos (QT >600 ms). Mutação, C. 3208 C > T (p. Q1070X) no gene KCNH2 foi encontrada nos doentes (mostrado à direita), marcados com uma seta.
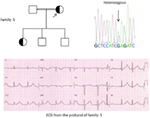
Figura 5. Superior esquerdo: pedigree da família-5. O indivíduo afetado é mostrado por círculo preenchido (feminino) Proband é indicado por uma seta. 12-lead ECG of the proband (i:2). O ECG mostra um alto pico, de base ampla, de grande amplitude da onda T, intervalo QTc é de 580 ms. Análise do gene KCNH2 mostra substituição do nucleotídeo “G” para “A” (c.2362G > A, seta marcada), o que leva ao aminoácido de substituição, p.E788K.
Genética clínica e achados em nosso estudo (12-14) da Arábia saudita são bastante intrigante, por várias razões: (1) No total, temos investigado seis famílias, entre eles, quatro eram homozigotas composto/heterozigotos para as mutações e as mutações que se originou a partir de um ancestral de origem; (2) todas as mutações em LQTS foram novas, relatadas apenas nessas famílias Árabes; (3) Devido à homozigosidade ou heterozigosidade composta para as mutações, os fenótipos clínicos também foram graves em nossas famílias estudadas (12-14). Sugerimos que as observações genéticas e fenotípicas provinham da extrema alta taxa de casamentos consanguíneos na Arábia Saudita (108, 109). Nosso estudo forneceu a primeira evidência científica sobre o papel da consanguinidade em exercer um papel fundamental em arritmias cardíacas inexplicáveis e Sdcs em crianças e adolescentes na Arábia Saudita (12-14). Também temos mostrado que a mutação KCNQ1 C. 387-5 T > a (NM_000218) (Figura 3) havia se espalhado na província Assir da Arábia Saudita a partir de um ancestral comum durante várias gerações devido à alta incidência de casamentos consanguíneos. Até agora, esta é a mutação causal LQTS1 mais comum na população da Arábia Saudita, que também foi observada no King Faisal Specialist Hospital e no Khamis Mashayt Military Hospital (Não publicado). Um resultado similar também foi obtido para a mutação causal LQTS2, C. 3208 C > T (p.Q1070X) (Figura 4) no gene KCNH2 (12, 14), que também é uma mutação dos fundadores na Arábia Saudita e segregada durante muitas gerações na região de Assir (veja o mapa, Figura 6). Seria de prever que existe um número considerável de indivíduos com o mencionado ancestral fundador mutações (tanto em KCNQ1 e KCNH2 e outros genes) nesta região e também em grandes cidades, como Riade, Jeddah e Dammam, devido à migração urbana. Mais mutações fundadoras ou ancestrais patogênicas aos LQTS são muito previstas em outras províncias da Arábia Saudita devido à alta taxa de casamentos consanguíneos.
Figura 6. Mapa da Arábia Saudita (cortesia: Wikipedia). A região de Assir é marcada com linha espessa, onde encontramos as mutações fundadoras nos genes KCNQ1 e KCNH2, descritas na família 1-4.
como o LQTS é uma doença autossómica dominante, esperávamos observar predominantemente doentes portadores de mutação heterozigótica. Mas em nosso estudo, identificamos principalmente pacientes com mutações recessivas (12-14). É evidente que estes pacientes se tornam os primeiros sintomas e, foram predominantemente encaminhados para o Prince Sultan Cardiac Centre, trazendo-os sob a nossa atenção. No entanto, os achados de nossas investigações implicam no fato de que os LQTS recessivos podem ter fenótipos clínicos fatais em crianças, e eles não são incomuns na Arábia Saudita (12-14). Como os portadores de mutação heterozigótica também são suscetíveis a desenvolver arritmia e suas complicações, uma iniciativa concertada deve ser tomada para trazer os médicos gerais locais, cardiologistas e geneticistas clínicos em uma plataforma comum para identificar os indivíduos em risco. Além disso, no momento, também não temos informação genética para outras doenças da arritmia familiar, por exemplo, CPVT, SQTS, BrS, AF, etc. Centros cardiogenéticos especializados devem tomar a iniciativa de procurar os defeitos genéticos, mutações, e realizar estudos genótipo-fenótipo em todas as formas de arritmias hereditárias. Deve-se também ter em consideração que nem todas as arritmias genéticas teriam uma história familiar, uma vez que, em muitos casos, as arritmias mutações causais são de novo na origem, ou seja, o proband é o primeiro paciente dessa família com a mutação e ele/ela é a fonte para transmitir a mutação em gerações a jusante (110, 111). Devido à alta taxa de casamentos consanguíneos na Arábia Saudita, esperamos que muito mais mutações de fundadores exercem um papel crucial em arritmias congênitas neste país. Identificar estas mutações fundadoras deve ser a nossa principal tarefa, o que nos facilitaria no desenvolvimento de um aconselhamento genético pré-matrimonial eficaz e também pré-sintomático neste país. Mutações em genes causais LQTS podem conferir variabilidade na penetração clínica, em um extremo, alguns portadores podem ser presumivelmente completamente saudáveis, mas alguns portadores podem ter sua primeira manifestação da doença como síncope ou morte súbita. A morte súbita de uma criança ou de um adulto representa uma grande carga psicológica e emocional para a família, a triagem dos portadores são essenciais, uma vez que existem medicamentos simples, por exemplo, β-bloqueadores (também modificação de comportamento), que poderiam muito efetivamente impedir os portadores das consequências fatais de arritmia e SCDs. Indivíduos com mutações homozigóticas nos genes causais do LQTS podem ter morbilidade grave e elevada taxa de mortalidade, incluindo brady-taquiarritmias fetais, e em muitos casos abortos espontâneos nas mães grávidas (12-14).
também não foram realizadas muitas pesquisas na Arábia Saudita sobre a prevalência de vários SNPs nos genes ligados à arritmia e também nos genes que os regulam. Splawski et al. (112) described a common S1103Y variant in the SCN5A gene associated with arritmia in African-Americans. A variante do alelo denominado Y1103 é responsável pela aceleração da ativação do canal, aumentando assim a probabilidade de arritmias cardíacas em pessoas de ascendência africana (52, 112). K393N é uma variante do gene KCNQ1 relatado em pacientes LQTS1 nos EUA, mas, na população árabe, detectamos esta variante em 2% dos indivíduos (dados não publicados). Se, variante K393N no gene Kcnq1 em árabes é comparável à variante S1103Y (SCN5A) em afro-americanos ou a variante D85N (KCNE1) na população japonesa também pode merecer investigação (59).
Declaração de conflito de interesses
os autores declaram que a investigação foi realizada na ausência de quaisquer relações comerciais ou financeiras que possam ser interpretadas como um potencial conflito de interesses.
agradecimentos
a nossa sincera gratidão ao Prof. Arthur Wilde, ao Prof. Connie Bezzina e à Editora da revista “Heart” pela sua permissão para usar a Figura 1 neste manuscrito da Ref. (113).
1. Bhuiyan ZA. Espectro clínico e genético de síndromes de arritmia cardíaca hereditária. Amsterdam: Amsterdam University (2009).
2. A Priori SG, Aliot e, Blømstrom-Lundqvist C, Bossaert L, Breithardt G, Brugada P, et al. Task force “morte súbita cardíaca”, sociedade europeia de Cardiologia. Europace (2002) 4:3-18. doi: 10.1053 / eupc.2001. 0214
CrossRef Texto Completo
3. Wang Q, Shen J, Splawski I, Atkinson D, Li Z, Robinson Jl, et al. Mutações SCN5A associadas a uma arritmia cardíaca hereditária, síndrome de QT longo. Cell (1995) 80:805-11. doi:10.1016/0092-8674(95)90359-3
Pubmed Resumo | Pubmed Texto Completo | CrossRef Texto Completo
4. Curran ME, Splawski I, Timothy KW, Vincent GM, Green ED, Keating MT. Uma base molecular para arritmia cardíaca: as mutações de HERG causam síndrome de QT longo. Cell (1995) 80:795-803. doi: 10.1016/0092-8674(95)90358-5
Pubmed Resumo | Pubmed Texto Completo | CrossRef Texto Completo
5. Wang Q, Curran ME, Splawski I, Burn TC, Millholland JM, VanRaay TJ, et al. Clonagem posicional de um novo gene do canal de potássio: mutações KVLQT1 causam arritmias cardíacas. Nat Genet (1996) 12:17-23. doi:10.1038/ng0196-17
Pubmed Resumo | Pubmed Texto Completo | CrossRef Texto Completo
6. Splawski I, Tristani-Firouzi M, Lehmann MH, Sanguinetti MC, Keating MT. Mutações no gene hminK causam síndrome de QT longo e suprimem a função IKs. Nat Genet (1997) 17:338-40. doi:10.1038/ng1197-338
Pubmed Resumo | Pubmed Texto Completo | CrossRef Texto Completo
7. Sanguinetti MC, Jiang C, Curran ME, Keating MT. Uma ligação mecanicista entre uma arritmia cardíaca herdada e adquirida: HERG codifica o canal de potássio IKr. Cell (1995) 81:299-307. doi:10.1016/0092-8674(95)90340-2
Pubmed Resumo | Pubmed Texto Completo | CrossRef Texto Completo
8. Neyroud N, Tesson F, Denjoy I, Leibovici M, Donger C, Barhanin J, et al. Uma nova mutação no gene KVLQT1 causa a síndrome cardioauditoria de Jervell e Lange-Nielsen. Nat Genet (1997) 15:186-9. doi:10.1038/ng0297-186
Pubmed Resumo | Pubmed Texto Completo | CrossRef Texto Completo
9. Kucera JP, Rohr s, Rudy Y. localização dos canais de sódio em discos intercalados modula condução cardíaca. Circ Res (2002) 91:1176-82. doi: 10.1161 / 01.RES.0000046237.54156.0 A
Pubmed Resumo | Pubmed Texto Completo | CrossRef Texto Completo
10. Em Oxford, Musa H, Maass K, Coombs W, Taffet SM, Delmar M. Connexin43 remodelação causada pela inibição da expressão da plakophilin-2 nas células cardíacas. Circ Res (2007) 101:703-11. doi: 10.1161 / CIRCRESAHA.107.154252
Pubmed Resumo | Pubmed Texto Completo | CrossRef Texto Completo
11. Rohr S. Molecular crosstalk entre junções mecânicas e elétricas no disco intercalado. Circ Res (2007) 101:637-9. doi: 10.1161 / CIRCRESAHA.107.161901
CrossRef Texto Completo
12. Bhuiyan ZA, Momenah TS, Gong Q, Amin AS, Ghamdi SA, Carvalho JS, et al. Perda intra-uterina recorrente devido à quase ausência de HERG: caracterização clínica e funcional de uma mutação homozigótica sem sentido HERG Q1070X. Heart Rhythm (2008) 5:553-61. doi: 10.1016 / j. hrthm.2008.01.020
Pubmed Resumo | Pubmed Texto Completo | CrossRef Texto Completo
13. Bhuiyan ZA, Momenah TS, Amin TS, Al-Khadra AS, Alders M, Wilde AAM, et al. Uma mutação Intrónica que leva a uma interrupção incompleta do exon-2 na KCNQ1 resgata a audição na síndrome de Jervell e Lange-Nielsen. Prog Biophys Mol Biol (2008) 98:319-27. doi: 10.1016 / j. pbiomolbio.2008.10.004
Pubmed Resumo | Pubmed Texto Completo | CrossRef Texto Completo
14. Bhuiyan ZA, Al-Shahrani S, Al-Khadra AS, Al-Ghamdi S, Al-Kalaf K, Mannens MMAM, et al. Análise clínica e genética da síndrome de QT longo em crianças de seis famílias na Arábia Saudita: são diferentes? Pediatr Cardiol (2009) 30:490-501. doi:10.1007/s00246-008-9377-y
Pubmed Resumo | Pubmed Texto Completo | CrossRef Texto Completo
15. Shinwari ZM, Al-Hazzani a, Dzimiri N, Tulbah S, Mallawi Y, Al-Fayyadh M, et al. Identificação de uma nova mutação do KCNQ1 numa grande família Saudita com síndrome de QT longo: consequências clínicas e implicações preventivas. Clin Genet (2013) 83:370-4. doi: 10.1111 / J. 1399-0004. 2012. 01914.X
Pubmed Abstract / Pubmed texto completo | CrossRef texto completo
16. Schwartz PJ, Stramba-Badiale M, Crotti L, Pedrazzini M, Besana a, Bosi G, et al. Prevalência da síndrome congénita de QT longo. Circulation (2009) 120:1761-7. doi: 10.1161 / CIRCULATIONAHA.109.863209
Pubmed Resumo | Pubmed Texto Completo | CrossRef Texto Completo
17. Schwartz PJ, Moss AJ, Vincent GM, Crampton RS. Critérios diagnósticos para a síndrome de QT longo. Actualizacao. Circulation (1993) 88:782-4. doi: 10.1161 / 01.CIR.88.2.782
CrossRef Texto Completo
18. A Priori SG, Wilde AA, Horie M, Cho Y, Behr ER, Berul C, et al. Resumo executivo: hrs/EHRA / APHRS consensus statement on the diagnosis and management of patients with inherited primary arritmia syndromes. Europace (2013) 15:1389-406. doi: 10.1093/europace / eut272
texto integral
19. Liu JF, Jons C, Moss AJ, McNitt s, Peterson DR, Qi M, et al. Registo De Síndrome. Factores de risco para síncope recorrente e acontecimentos fatais ou quase fatais subsequentes em crianças e adolescentes com síndrome de QT longo. J Am Coll Cardiol (2011) 57:941-50. doi: 10.1016 / j. jacc.2010.10.025
Pubmed Resumo | Pubmed Texto Completo | CrossRef Texto Completo
20. Splawski I, Shen J, Timothy KW, Lehmann MH, a Priori S, Robinson Jl, et al. Espectro de mutações nos genes da síndrome de QT longo. KVLQT1, HERG, SCN5A, KCNE1, e KCNE2. Circulation (2000) 102:1178-85. doi: 10.1161 / 01.CIR.102.10.1178
Pubmed Resumo | Pubmed Texto Completo | CrossRef Texto Completo
21. Westenskow P, Splawski I, Timothy KW, Keating MT, Sanguinetti MC. Mutações compostas: uma causa comum de síndrome de QT longo grave. Circulation (2004) 109:1834-41. doi: 10.1161 / 01.CIR.0000125524.34234.13
Pubmed Resumo | Pubmed Texto Completo | CrossRef Texto Completo
22. Mullally J, Goldenberg I, Moss AJ, Lopes CM, Ackerman MJ, Zareba W, et al. Risco de acontecimentos cardíacos com risco de vida em doentes com síndrome de QT longo e mutações múltiplas. Heart Rhythm (2013) 10:378-82. doi: 10.1016 / j. hrthm.2012.11.006
Pubmed Resumo | Pubmed Texto Completo | CrossRef Texto Completo
23. Napolitano C, A Priori SG, Schwartz PJ, Bloise R, Ronchetti e, Nastoli J, et al. Genetic testing in the long QT syndrome: development and validation of an efficient approach to genotyping in clinical practice. JAMA (2005) 294:2975-80. doi: 10.1001 / jama.294.23.2975
Pubmed Resumo | Pubmed Texto Completo | CrossRef Texto Completo
24. Roden DM. Clinico. Síndrome de QT longo. N Engl J Med (2008) 358:169-76. doi:10.1056/NEJMcp0706513
CrossRef Texto Completo
25. Schwartz PJ, a Priori SG, Spazzolini C, Moss AJ, Vincent GM, Napolitano C, et al. Correlação genótipo-fenótipo na síndrome do intervalo QT longo: desencadeadores específicos do gene para arritmias com risco de vida. Circulation (2001) 103:89-95. doi: 10.1161 / 01.CIR.103.1.89
CrossRef Texto Completo
26. Ackerman MJ, Tester DJ, Porter CJ. Natação, um detonador arritmogénico específico de um gene para a síndrome de QT longo hereditário. Mayo Clin Proc (1999) 74:1088-94. doi:10.4065/74.11.1088
Pubmed Resumo | Pubmed Texto Completo | CrossRef Texto Completo
27. Kapa S, Tester DJ, Salisbury BA, Harris-Kerr C, Pungliya MS, Alders M, et al. Ensaios genéticos para a síndrome de QT longo: distinguindo mutações patogénicas das variantes benignas. Circulation (2009) 120:1752-60. doi: 10.1161 / CIRCULATIONAHA.109.863076
Pubmed Resumo | Pubmed Texto Completo | CrossRef Texto Completo
28. Moss AJ, Shimizu W, Wilde AA, Towbin JA, Zareba W, Robinson Jl, et al. Aspectos clínicos da síndrome de Long-QT tipo 1 por localização, tipo de codificação e função Biofísica das mutações que envolvem o gene KCNQ1. Circulation (2007) 115:2481-9. doi: 10.1161 / CIRCULATIONAHA.106.665406
Pubmed Resumo | Pubmed Texto Completo | CrossRef Texto Completo
29. Amin AS, Giudicessi JR, Tijsen AJ, Spanjaart AM, Reckman YJ, Klemens CA, et al. As variantes na Região 3 ‘ não traduzida do canal de potássio kcnq1 codificado Kv7.1 modificam a gravidade da doença em doentes com síndrome de QT longo tipo 1 de uma forma alélica específica. Eur Heart J (2012) 33:714-23. doi:10.1093/eurheartj/ehr473
Pubmed Resumo | Pubmed Texto Completo | CrossRef Texto Completo
30. Barsheshet a, Goldenberg I, O-Uchi J, Moss AJ, Jons C, Shimizu W, et al. Mutações nos lacetes citoplásmicos do canal KCNQ1 e risco de acontecimentos com risco de vida: implicações para a resposta específica à terapêutica com bloqueadores β na síndrome de QT longo tipo 1. Circulation (2012) 125:1988-96. doi: 10.1161 / CIRCULATIONAHA.111.048041
Pubmed Resumo | Pubmed Texto Completo | CrossRef Texto Completo
31. Schwartz PJ, Spazzolini C, Crotti l, Bathen J, Amlie JP, Timothy K, et al. The Jervell and Lange-Nielsen syndrome: natural history, molecular basis, and clinical outcome. Circulation (2006) 113:783-90. doi: 10.1161 / CIRCULATIONAHA.105.592899
Pubmed Resumo | Pubmed Texto Completo | CrossRef Texto Completo
32. Bhuiyan ZA, Wilde AA. Com o coração e a audição, a orelha precisa de menos do que o coração. Circ Cardiovasc Genet (2013) 6:141-3. doi: 10.1161 / CIRCGENETICS.113.000143
CrossRef Texto Completo
33. Wollnik B, Schroeder BC, Kubisch C, Esperer HD, Wieacker P, Jentsch TJ. Mecanismos patofisiológicos de mutações do canal kvlqt1 k+ dominantes e recessivas encontradas em arritmias cardíacas hereditárias. Hum Mol Genet (1997) 6:1943-9. doi: 10.1093 / hmg / 6.11.1943
CrossRef Texto Completo
34. Wilde AA, Jongbloed RJ, Doevendans PA, Düren DR, Hauer RN, van Langen IM, et al. Estímulos auditivos como um gatilho para eventos arrítmicos diferenciam pacientes relacionados com HERG (LQTS2) de pacientes relacionados com KVLQT1 (LQTS1). J Am Coll Cardiol (1999) 33:327-32. doi:10.1016/S0735-1097(98)00578-6
CrossRef Texto Completo
35. Moss AJ, Zareba W, Kaufman ES, Gartman e, Peterson DR, Benhorin J, et al. Aumento do risco de acontecimentos arrítmicos na síndrome de QT longo com mutações na região dos poros do canal de potássio genético relacionado com o éter-a-go-humano. Circulation (2002) 105:794-9. doi:10.1161/hc0702.105124
Pubmed Resumo | Pubmed Texto Completo | CrossRef Texto Completo
36. Lupoglazoff JM, Denjoy I, Villain e, Fressart V, Simon F, Bozio A, et al. Síndrome de QT longo em recém-nascidos: perturbações da condução associadas a mutações HERG e bradicardia sinusal com mutações KCNQ1. J Am Coll Cardiol (2004) 43:826-30. doi: 10.1016 / j. jacc.2003.09.049
Pubmed Resumo | Pubmed Texto Completo | CrossRef Texto Completo
37. Chevalier P, Bellocq C, Millat G, Piqueras E, Potet F, Schott JJ, et al. Torsades de pointes complicando o bloqueio atrioventricular: evidência para uma predisposição genética. Heart Rhythm (2007) 4:170-4. doi: 10.1016 / j. hrthm.2006.10.004
Pubmed Resumo | Pubmed Texto Completo | CrossRef Texto Completo
38. Hoorntje T, Alders M, van Tintelen P, van der Lip K, Sreeram N, van der Wal a, et al. Truncação homozigótica prematura da proteína HERG: o knockout humano HERG. Circulation (1999) 100:1264-7. doi: 10.1161 / 01.CIR.100.12.1264
Pubmed Resumo | Pubmed Texto Completo | CrossRef Texto Completo
39. Pippo K, Laitinen P, Swan H, Toivonen L, Viitasalo M, Pasternack M, et al. A homozigosidade de uma mutação do canal de potássio HERG causa uma forma grave de síndrome de QT longo: identificação de uma aparente mutação fundadora nos finlandeses. J Am Coll Cardiol (2000) 35:1919-25. doi:10.1016/S0735-1097(00)00636-7
Pubmed Resumo | Pubmed Texto Completo | CrossRef Texto Completo
40. Johnson WHR, Yang P, Yang T, Lau YR, Mostella BA, Wolff DJ, et al. Caracterização clínica, genética e Biofísica de uma mutação HERG homozigosa causando grave síndrome neonatal de QT longo. Pediatr Res (2003) 53:744-8. doi: 10.1203 / 01.PDR.0000059750.17002.B6
Pubmed Abstract / Pubmed Texto Completo | CrossRef Texto Completo
41. Ackerman MJ, Tester DJ, Jones GS, Will ML, Burrow CR, Curran ME. Diferenças étnicas nas variantes do canal cardíaco de potássio: implicações para a susceptibilidade genética a morte súbita cardíaca e testes genéticos para a síndrome de QT longo congénito. Mayo Clin Proc (2003) 78:1479-87. doi:10.4065/78.12.1479
Pubmed Resumo | Pubmed Texto Completo | CrossRef Texto Completo
42. Crotti L, Lundquist AL, Insolia R, Pedrazzini M, Ferrandi C, De Ferrari GM, et al. KCNH2-K897T é um modificador genético da síndrome congênita de QT longo. Circulation (2005) 112:1251-8. doi: 10.1161 / CIRCULATIONAHA.105.549071
Pubmed Resumo | Pubmed Texto Completo | CrossRef Texto Completo
43. Nof e, Cordeiro JM, Pérez GJ, Scornik FS, Calloe K, Love B, et al. Um polimorfismo simples de nucleótidos pode exacerbar a síndrome do tipo 2 do QT longo, levando à morte súbita do bebé. Circ Cardiovasc Genet (2010) 3:199-206. doi: 10.1161 / CIRCGENETICS.109.898569
Pubmed Resumo | Pubmed Texto Completo | CrossRef Texto Completo
44. Bezzina C, Veldkamp MW, van Den Berg MP, Postma AV, Rook MB, Viersma JW, et al. Uma mutação única do canal Na (+) causando síndromes de longo QT e Brugada. Circ Res (1999) 85:1206-13. doi: 10.1161 / 01.RES.85.12.1206
Pubmed Resumo | Pubmed Texto Completo | CrossRef Texto Completo
45. Kyndt F, Probst V, Potet F, Demolombe S, Chevallier JC, Baro i, et al. Nova mutação SCN5A que conduz a um defeito isolado de condução cardíaca ou síndrome de Brugada numa grande família Francesa. Circulation (2001) 104:3081-6. doi:10.1161/hc5001.100834
Pubmed Resumo | Pubmed Texto Completo | CrossRef Texto Completo
46. Makita N, Behr e, Shimizu W, Horie M, Sunami a, Crotti L, et al. A mutação e1784k na SCN5A está associada a fenótipo clínico misto da síndrome de QT longo tipo 3. J Clin Invest (2008) 118:2219-29. doi:10.1172/JCI34057
Pubmed Resumo | Pubmed Texto Completo | CrossRef Texto Completo
47. Keller DI, Acharfi S, Delacrétaz E, Benammar N, Rotter M, Pfammatter JP, et al. Uma nova mutação no SCN5A, delQKP 1507-1509, causando síndrome de QT longo: papel do resíduo Q1507 na inactivação do canal de sódio. J Mol Cell Cardiol (2003) 35:1513-21. doi: 10.1016 / j. yjmcc.2003.08.007
Pubmed Resumo | Pubmed Texto Completo | CrossRef Texto Completo
48. A Priori SG, Schwartz PJ, Napolitano C, Bloise R, Ronchetti E, Grillo M, et al. Estratificação do risco na síndrome de QT longo. N Engl J Med (2003) 348:1866-74. doi:10.1056/NEJMoa022147
Pubmed Resumo | Pubmed Texto Completo | CrossRef Texto Completo
49. Lupoglazoff JM, Cheav T, Baroudi G, Berthet M, Denjoy I, Cauchemez B, et al. Mutação da SCN5A homozigótica na síndrome de QT longo com bloqueio auriculoventricular funcional de dois a um. Circ Res (2001) 89:E16–21. doi: 10.1161/hh1401.095087
CrossRef Texto Completo
50. Shi R, Zhang Y, Yang C, Huang C, Zhou X, Qiang H, et al. A mutação do canal cardíaco de sódio delQKP 1507-1509 está associada ao espectro fenotípico em expansão do LQT3, desordem de condução, cardiomiopatia dilatada e elevada incidência de morte súbita juvenil. Europace (2008) 10:1329-35. doi:10.1093/europace/eun202
Pubmed Resumo | Pubmed Texto Completo | CrossRef Texto Completo
51. Zumhagen S, Veldkamp MW, Stallmeyer B, Baartscheer a, Eckardt L, Paul M, et al. Uma mutação heterozigótica por eliminação no gene SCN5A do canal cardíaco de sódio com características de perda e ganho de função manifesta – se como doença de condução isolada, sem sinais de Brugada ou síndrome de QT longo. PLoS One (2013) 8:e67963. doi: 10.1371 / journal.pone.0067963
Pubmed Resumo | Pubmed Texto Completo | CrossRef Texto Completo
52. Plant LD, Bowers PN, Liu Q, Morgan T, Zhang T, State MW, et al. A common cardiac sodium channel variant associated with sudden infant death in African Americans, SCN5A S1103Y. J Clin Invest (2006) 116:430-5. doi: 10.1172/JCI25618
Pubmed Resumo | Pubmed Texto Completo | CrossRef Texto Completo
53. Mohler PJ, Schott JJ, Gramolini AO, Dilly KW, Guatimosim S, duBell WH, et al. A mutação anquirina-B causa arritmia cardíaca de tipo 4 de intervalo QT longo e morte cardíaca súbita. Nature (2003) 421:634-9. doi:10.1038/nature01335
Pubmed Resumo | Pubmed Texto Completo | CrossRef Texto Completo
54. Schott JJ, Charpentier F, Peltier S, Foley P, Drouin e, Bouhour JB, et al. Mapeamento de um gene para a síndrome de QT longo para o cromossoma 4q25-27. Am J Hum Genet (1995) 57:1114-22.
Pubmed Abstract / Pubmed Texto Completo
55. Barhanin J, Lesage F, Guillemare e, Fink M, Lazdunski M, Romey G. K(V)lqt1 e proteínas lsK (minK) associam-se para formar a corrente I(Ks) de potássio cardíaco. Nature (1996) 384:78-80. doi:10.1038/384078a0
Pubmed Resumo | Pubmed Texto Completo | CrossRef Texto Completo
56. Sanguinetti MC, Curran ME, Zou a, Shen J, Spector PS, Atkinson DL, et al. Coassembly of K (V)LQT1 and minK(IsK) proteins to form cardiac I (Ks) potassium channel. Nature (1996) 384:80-3. doi: 10.1038/384080a0
Pubmed Resumo | Pubmed Texto Completo | CrossRef Texto Completo
57. Schulze-Bahr e, Wang Q, Wedekind H, Haverkamp W, Chen Q, Sun Y, et al. Mutações KCNE1 causam Jervell e síndrome Lange-Nielsen. Nat Genet (1997) 17:267-8. doi:10.1038/ng1197-267
CrossRef Texto Completo
58. Duggal P, Vesely MR, Wattanasirichaigoon D, Villafane J, Kaushik V, Beggs AH. Mutação do gene para a IKs associada tanto a Jervell como A Lange-Nielsen e a Romano-Ward formas de síndrome de QT longo. Circulation (1998) 97:142-6. doi: 10.1186/1471-2350-9-24
Pubmed Resumo | Pubmed Texto Completo | CrossRef Texto Completo
59. Nishio Y, Makiyama T, Itoh H, Sakaguchi T, Ohno S, Gong YZ, et al. D85N, um polimorfismo KCNE1, é uma variante genética causadora de doenças na síndrome de QT longo. J Am Coll Cardiol (2009) 54:812-9. doi: 10.1016 / j. jacc.2009.06.005
Pubmed Resumo | Pubmed Texto Completo | CrossRef Texto Completo
60. Paulussen AD, Gilissen RA, Armstrong m, Doevendans PA, Verhasselt P, Smeets HJ, et al. Variações genéticas de KCNQ1, KCNH2, SCN5A, KCNE1 e KCNE2 em doentes com síndrome de QT longo induzido por fármacos. Mol Med (2004) 82:182-8. doi:10.1007/s00109-003-0522-z
Pubmed Resumo | Pubmed Texto Completo | CrossRef Texto Completo
61. Abbott GW, Sesti F, Splawski I, Buck ME, Lehmann MH, Timothy KW, et al. MiRP1 forma canais IKr de potássio com HERG e está associado com arritmia cardíaca. Cell (1999) 97:175-87. doi:10.1016/S0092-8674(00)80728-X
Pubmed Resumo | Pubmed Texto Completo | CrossRef Texto Completo
62. Gordon e, Panaghie G, Deng l, Bee KJ, Roepke TK, Krogh-Madsen T, et al. Uma mutação KCNE2 num doente com arritmia cardíaca induzida por estímulos auditivos e desequilíbrio electrolítico sérico. Cardiovasc Res (2008) 77:98-106. doi:10.1093/cvr/cvm030
Pubmed Resumo | Pubmed Texto Completo | CrossRef Texto Completo
63. Gesso NM, Tawil R, Tristani-Firouzi M, Canún S, Bendahhou S, Tsunoda A, et al. Mutações em Kir2. 1 causam o desenvolvimento e fenótipos elétricos episódicos da síndrome de Andersen. Cell (2001) 105:511-9. doi: 10.1016/S0092-8674(01)00342-7
Pubmed Resumo | Pubmed Texto Completo | CrossRef Texto Completo
64. Kubo Y, Baldwin TJ, Jan YN, Jan LY. Estrutura primária e expressão funcional de um canal de potássio retificador interno do rato. Nature (1993) 362:127-33. doi:10.1038/362127a0
Pubmed Resumo | Pubmed Texto Completo | CrossRef Texto Completo
65. Raab-Graham KF, Radeke CM, Vandenberg CA. Clonagem Molecular e expressão de um coração humano no interior do canal de potássio retificador. Neurorreport (1994) 5:2501-5. doi: 10.1097/00001756-199412000-00024
Pubmed Resumo | Pubmed Texto Completo | CrossRef Texto Completo
66. Splawski I, Timothy KW, Sharpe LM, Decher N, Kumar P, Bloise R, et al. A disfunção dos canais de cálcio ca (V)1, 2 causa uma perturbação multisistema, incluindo arritmia e autismo. Cell (2004) 119:19-31. doi: 10.1016/j. cell.2004.09.011
Pubmed Resumo | Pubmed Texto Completo | CrossRef Texto Completo
67. Splawski I, Timothy KW, Decher N, Kumar P, Sachse FB, Beggs AH, et al. Perturbação grave da arritmia causada por mutações cardíacas dos canais de cálcio tipo L. Proc Natl Acad Sci U S A (2005) 102:8089-96. doi: 10.1073 / pnas.0502506102
Pubmed Resumo | Pubmed Texto Completo | CrossRef Texto Completo
68. Gillis J, Burashnikov E, Antzelevitch C, Blaser S, Gross G, Turner L, et al. Longo intervalo QT, sindactilia, contracturas articulares, acidente vascular cerebral e mutação Nova CACNA1C: expansão do espectro da síndrome de Timóteo. Am J Med Genet a (2012) 158A:182-7. doi: 10.1002 / ajmg.a.34355
Pubmed Resumo | Pubmed Texto Completo | CrossRef Texto Completo
69. Dufendach KA, Giudicessi JR, Boczek NJ, Ackerman MJ. O mosaicismo materno confunde o diagnóstico neonatal da síndrome de Timóteo tipo 1. Pediatria (2013) 131:e1991–5. doi: 10.1542 / Pediatria.2012-2941
Pubmed Resumo | Pubmed Texto Completo | CrossRef Texto Completo
70. Vatta M, Ackerman MJ, Ye B, Makielski JC, Ughanze EE, Taylor EW, et al. O mutante caveolin-3 induz uma corrente de sódio tardia persistente e está associado à síndrome de QT longo. Circulação (2006) 114:2104–12. doi: 10.1161 / CIRCULATIONAHA.106.635268
Pubmed Resumo | Pubmed Texto Completo | CrossRef Texto Completo
71. Cronk LB, Ye B, Kaku T, Tester DJ, Vatta M, Makielski JC, et al. Mecanismo novo para a síndrome de morte súbita infantil: corrente de sódio tardia persistente secundária a mutações na caveolin-3. Heart Rhythm (2007) 4:161-6. doi: 10.1016 / j. hrthm.2006.11.030
Pubmed Resumo | Pubmed Texto Completo | CrossRef Texto Completo
72. Engelman JA, Zhang X, Galbiati F, Volonte D, Sotgia F, Pestell RG, et al. Molecular genetics of the caveolin gene family: implications for human cancers, diabetes, Alzheimer disease, and muscular distrophy. Am J Hum Genet (1998) 63:1578-87. doi:10.1086/302172
CrossRef Texto Completo
73. Balijepalli RC, Foell JD, Hall DD, Hell JW, Kamp TJ. Para a regulação beta-adrenérgica é necessária a localização dos canais cardíacos L-tipo Ca(2+) para um complexo de sinalização macromolecular caveolar. Proc Natl Acad Sci U S A (2006) 103:7500-5. doi: 10.1073 / pnas.0503465103
Pubmed Resumo | Pubmed Texto Completo | CrossRef Texto Completo
74. Medeiros-Domingo a, Kaku T, Tester DJ, Iturralde-Torres P, Itty a, Ye B, et al. Subunidade do canal de sódio codificado com SCN4B na síndrome de QT longo congénito. Circulation (2007) 116:134-42. doi: 10.1161 / CIRCULATIONAHA.106.659086
Pubmed Resumo | Pubmed Texto Completo | CrossRef Texto Completo
75. Chen L, Marquardt ML, Tester DJ, Sampson KJ, Ackerman MJ, Kass RS. A mutação de uma proteína de Ancoragem a-kinase causa síndrome de QT longo. Proc Natl Acad Sci U S A (2007) 104:20990-5. doi: 10.1073 / pnas.0710527105
Pubmed Resumo | Pubmed Texto Completo | CrossRef Texto Completo
76. Wu G, Ai T, Kim JJ, Mohapatra B, Xi Y, Li Z, et al. Mutação alfa-1-sintrofina e Síndrome de QT longo: uma doença de perturbação do canal de sódio. Circ Arrhythm Electrophysiol (2008) 1:193-201. doi: 10.1161 / CIRCEP.108.769224
Pubmed Resumo | Pubmed Texto Completo | CrossRef Texto Completo
77. Yang Y, Yang Y, Liang B, Liu J, Li J, Grunnet M, et al. Identificação de um Kir3.Mutação na síndrome de QT longo congénito. Am J Hum Genet (2010) 86:872-80. doi: 10.1016 / j. ajhg.2010.04.017
Pubmed Resumo | Pubmed Texto Completo | CrossRef Texto Completo
78. Roden DM. Mecanismos e gestão de proarritmia. Am J Cardiol (1998) 82:49I–57I. doi:10.1016/S0002-9149(98)00472-X
CrossRef Texto Completo
79. Mitcheson JS, Chen J, Lin M, Culberson c, Sanguinetti MC. Uma base estrutural para a síndrome de QT longo induzida por fármacos. Proc Natl Acad Sci U S A (2000) 97:12329-33. doi: 10.1073 / pnas.210244497
CrossRef Texto Completo
80. Kannankeril PJ, Roden DM. QT longo induzido por drogas e torsade de pointes: avanços recentes. Curr Opin Cardiol (2007) 22:39-43. doi: 10.1097 / HCO.0b013e32801129eb
Pubmed Resumo | Pubmed Texto Completo | CrossRef Texto Completo
81. Veerman CC, Verkerk AO, Blom MT, Klemens CA, Langendijk PN, van Ginneken AC, et al. Bloqueio retificador lento retardado de potássio contribui de forma importante para a síndrome de QT longo induzido pelo fármaco. Circ Arrhythm Electrophysiol (2013) 6:1002-9. doi: 10.1161 / CIRCEP.113.000239
Pubmed Resumo | Pubmed Texto Completo | CrossRef Texto Completo
82. Sesti F, Abbott GW, Wei J, Murray KT, Saksena S, Schwartz PJ, et al. Um polimorfismo comum associado a arritmia cardíaca induzida por antibióticos. Proc Natl Acad Sci U S A (2000) 97:10613-8. doi: 10.1073 / pnas.180223197
Pubmed Resumo | Pubmed Texto Completo | CrossRef Texto Completo
83. Kääb S, Crawford DC, Sinner MF, Behr ER, Kannankeril PJ, Wilde AA, et al. A large candidate gene survey identifies the KCNE1 D85N polymorphism as a possible modulator of drug-induced torsades de pointes. Circ Cardiovasc Genet (2012) 5:91-9. doi: 10.1161 / CIRCGENETICS.111.960930
Pubmed Resumo | Pubmed Texto Completo | CrossRef Texto Completo
84. Nakamura K, Katayama Y, Kusano KF, Haraoka K, Tani Y, Nagase S, et al. Síndrome de QT longo induzido por anticorpos Anti-KCNH2: nova forma adquirida de síndrome de QT longo. J Am Coll Cardiol (2007) 50:1808-9. doi: 10.1016 / j. jacc.2007.07.037
CrossRef Texto Completo
85. Moss AJ, Zareba W, Benhorin J, Locati EH, Hall WJ, Robinson Jl, et al. Padrões de ondas T de ECG em formas geneticamente distintas da síndrome de QT longo hereditário. Circulation (1995) 92:2929-34. doi: 10.1161 / 01.CIR.92.10.2929
Pubmed Resumo | Pubmed Texto Completo | CrossRef Texto Completo
86. Zhang L, Timothy KW, Vincent GM, Lehmann MH, Fox J, Giuli LC, et al. Espectro de padrões de onda ST-T e parâmetros de repolarização na síndrome congénita de longo intervalo QT: os resultados do ECG identificam genótipos. Circulation (2000) 102:2849-55. doi: 10.1161 / 01.CIR.102.23.2849
Pubmed Resumo | Pubmed Texto Completo | CrossRef Texto Completo
87. Tan HL, Bardai a, Shimizu W, Moss AJ, Schulze-Bahr e, Noda T, et al. Aparecimento específico do genótipo de arritmias na síndrome de QT longo congénito: possíveis implicações da terapêutica. Circulation (2006) 114:2096-103. doi: 10.1161 / CIRCULATIONAHA.106.642694
Pubmed Resumo | Pubmed Texto Completo | CrossRef Texto Completo
88. Locati EH, Zareba W, Moss AJ, Schwartz PJ, Vincent GM, Lehmann MH, et al. Diferenças de manifestações clínicas relacionadas com a idade e o Sexo em doentes com síndrome de QT longo congénito: resultados do Registo Internacional de LQTS. Circulation (1998) 97:2237-44. doi: 10.1161 / 01.CIR.97.22.2237
Pubmed Resumo | Pubmed Texto Completo | CrossRef Texto Completo
89. Zareba W, Moss AJ, Schwartz PJ, Vincent GM, Robinson Jl, Priori SG, et al. Influência do genótipo no curso clínico do síndrome de QT longo. International Long-QT Syndrome Registry Research Group. N Engl J Med (1998) 339:960-5. doi: 10.1056/NEJM199810013391404
Pubmed Resumo | Pubmed Texto Completo | CrossRef Texto Completo
90. Goldenberg I, Moss AJ, Bradley J, Polonsky S, Peterson DR, McNitt s, et al. Síndrome de QT longo após os 40 anos de idade. Circulation (2008) 117:2192-201. doi: 10.1161 / CIRCULATIONAHA.107.729368
Pubmed Resumo | Pubmed Texto Completo | CrossRef Texto Completo
91. Zareba W, Moss AJ, Locati EH, Lehmann MH, Peterson DR, Hall WJ, et al. Registo De Síndrome. Modular os efeitos da idade e do sexo no decurso clínico da síndrome de QT longo por genótipo. J Am Coll Cardiol (2003) 42:103-9. doi:10.1016/S0735-1097(03)00554-0
Pubmed Resumo | Pubmed Texto Completo | CrossRef Texto Completo
92. Seth R, Moss AJ, McNitt s, Zareba W, Andrews ML, Qi M, et al. Síndrome de QT longo e gravidez. J Am Coll Cardiol (2007) 49:1092-8. doi: 10.1016 / j. jacc.2006.09.054
Pubmed Resumo | Pubmed Texto Completo | CrossRef Texto Completo
93. Schwartz PJ, a Priori SG, Dumaine R, Napolitano C, Antzelevitch C, Stramba-Badiale M, et al. Uma ligação molecular entre a síndrome de morte súbita infantil e a síndrome de QT longo. N Engl J Med (2000) 343:262-7. doi:10.1056/NEJM200007273430405
CrossRef Texto Completo
94. Schwartz PJ, a Priori SG, Bloise R, Napolitano C, Ronchetti E, Piccinini A, et al. Diagnóstico Molecular numa criança com síndrome de morte súbita infantil. Lancet (2001) 358:1342-3. doi:10.1016/S0140-6736(01)06450-9
Pubmed Resumo | Pubmed Texto Completo | CrossRef Texto Completo
95. Christiansen M, Tønder N, Larsen LA, Andersen PS, Simonsen H, Oyen N, et al. Mutations in the HERG K+ – ion channel: a novel link between long QT syndrome and sudden infant death syndrome. Am J Cardiol (2005) 95:433-4. doi: 10.1016 / j. amjcard.2004.09.054
Pubmed Resumo | Pubmed Texto Completo | CrossRef Texto Completo
96. Tester DJ, Ackerman MJ. Síndrome de QT longo pós-morte prolongada teste genético para morte súbita inexplicável nos jovens. J Am Coll Cardiol (2007) 49:240-6. doi: 10.1016 / j. jacc.2006.10.010
Pubmed Resumo | Pubmed Texto Completo | CrossRef Texto Completo
97. Hofman N, Tan HL, Clur SA, Alders M, van Langen IM, Wilde AA. Contribuição de doença cardíaca hereditária para morte cardíaca súbita na infância. Pediatria (2007) 120:e967–73. doi: 10.1542 / Pediatria.2006-3751
Pubmed Resumo | Pubmed Texto Completo | CrossRef Texto Completo
98. Wedekind H, Smits JP, Schulze-Bahr e, Arnold R, Veldkamp MW, Bajanowski T, et al. Mutação de novo no gene SCN5A associada ao início precoce da morte súbita do bebé. Circulation (2001) 104:1158-64. doi:10.1161/hc3501.095361
CrossRef Texto Completo
99. Wang DW, Desai RR, Crotti L, Arnestad M, Insolia R, Pedrazzini M, et al. Disfunção do canal de sódio cardíaco na síndrome de morte súbita infantil. Circulation (2007) 115:368-76. doi: 10.1161 / CIRCULATIONAHA.106.646513
Pubmed Resumo | Pubmed Texto Completo | CrossRef Texto Completo
100. Van Norstrand DW, Valdivia CR, Tester DJ, Ueda K, London B, Makielski JC, et al. Caracterização Molecular e funcional de novas mutações do gene do tipo glicerol-3-fosfato desidrogenase 1 (GPD1-L) na síndrome de morte súbita infantil. Circulation (2007) 116:2253-9. doi: 10.1161 / CIRCULATIONAHA.107.704627
Pubmed Resumo | Pubmed Texto Completo | CrossRef Texto Completo
101. Tan BH, Pundi KN, Van Norstrand DW, Valdivia CR, Tester DJ, Medeiros-Domingo A, et al. Mutações associadas à síndrome da Morte Súbita Infantil nas subunidades beta do canal de sódio. Heart Rhythm (2010) 7:771-8. doi: 10.1016 / j. hrthm.2010.01.032
Pubmed Resumo | Pubmed Texto Completo | CrossRef Texto Completo
102. Miller TE, Estrella E, Myerburg RJ, Garcia de Viera J, Moreno N, Rusconi P, et al. Perda fetal recorrente do terceiro trimestre e mosaicismo materno para o síndrome de QT longo. Circulation (2004) 109:3029-34. doi: 10.1161 / 01.CIR.0000130666.81539.9 E
Pubmed Resumo | Pubmed Texto Completo | CrossRef Texto Completo
103. Chockalingam P, Crotti L, Girardengo G, Johnson JN, Harris KM, van der Heijden JF, et al. Nem todos os beta-bloqueadores são iguais no tratamento do síndrome de QT longo tipos 1 e 2: recorrências mais elevadas de eventos sob Metoprolol. J Am Coll Cardiol (2012) 60:2092-6. doi: 10.1016 / j. jacc.2012.07.046
Pubmed Abstract / Pubmed Texto Completo | CrossRef Texto Completo
104. Schwartz PJ, a Priori SG, Cerrone M, Spazzolini C, Odero a, Napolitano C, et al. Denervação por simpatia cardíaca esquerda no tratamento de doentes de alto risco afectados pela síndrome de QT longo. Circulation (2004) 109:1826-33. doi: 10.1161 / 01.CIR.0000125523.14403.1 E
Pubmed Resumo | Pubmed Texto Completo | CrossRef Texto Completo
105. Singh B, al Shahwan SA, Habbab MA, al Deeb SM, biary N. Idiopathic long QT syndrome: asking the right question. Lancet (1993) 341:741. doi:10.1016/0140-6736(93)90501-7
CrossRef Texto Completo
106. Gorgels AP, Al Fadley F, Zaman L, Kantoch MJ, Al Halees Z. síndrome de QT longo com condução auriculoventricular diminuída: uma variante maligna em crianças. J Cardiovasc Electrophysiol (1998) 9:1225-32. doi: 10.1111 / J. 1540-8167. 1998.tb00096.X
Pubmed Abstract / Pubmed texto completo | CrossRef texto completo
107. Kantoch MJ, Qurashi MM, Bulbul ZR, Gorgels AP. Um recém-nascido com uma doença cardíaca congénita complexa, bloqueio auriculoventricular e taquicardia ventricular torsade de pointes. Pacing Clin Electrophysiol (1998) 21:2664-7. doi: 10.1111 / J. 1540-8159. 1998.tb00043.X
CrossRef texto completo
108. El-Hazmi MA, al-Swailem AR, Warsy AS, al-Swailem AM, Sulaimani R, al-Meshari AA. Consanguinidade entre a população da Arábia Saudita. J Med Genet (1995) 32:623-6. doi: 10.1136 / jmg.32. 8. 623
Texto Integral
109. El Mouzan MI, Al Salloum AA, Al Herbish AS, Qurachi MM, Al Omar AA. Consanguinidade e grandes distúrbios genéticos nas crianças Sauditas: um estudo transversal baseado na comunidade. Ann Saudi Med (2008) 28:169-73. doi:10.4103/0256-4947.51726
Pubmed Resumo | Pubmed Texto Completo | CrossRef Texto Completo
110. Medeiros-Domingo a, Bhuiyan ZA, Tester DJ, Hofman N, Bikker H, van Tintelen JP, et al. O canal de libertação do receptor / cálcio codificado de RYR2 em doentes diagnosticados previamente com taquicardia polimórfica polimórfica polimórfica polimórfica ou com síndrome QT longo, induzido pelo exercício, negativo: a comprehensive open reading frame mutational analysis. J Am Coll Cardiol (2009) 54:2065-74. doi: 10.1016 / j. jacc.2009.08.022
Pubmed Resumo | Pubmed Texto Completo | CrossRef Texto Completo
111. Al-Aama JY, Al-Ghamdi S, Bdier AY, Wilde AA, Bhuiyan ZA. Mutação de novo no gene KCNQ1 causal a Jervell e síndrome Lange-Nielsen. Clin Genet (2013). doi: 10.1111/cge.12300
Pubmed Abstract / Pubmed Texto Completo | CrossRef Texto Completo
112. Splawski I, Timothy KW, Tateyama M, Clancy CE, Malhotra A, Beggs AH, et al. Variante do canal de sódio SCN5A implicada no risco de arritmia cardíaca. Science (2002) 297:1333-6. doi: 10.1126 / science.1073569
CrossRef Texto Completo
113. Wilde AA, Bezzina CR. Genética de arritmias cardíacas. Heart (2005) 91:1352-8.