Mucopolysaccharidoses (MPS) são um grupo de clinicamente heterogêneos doenças causadas por deficiências do lysosomal enzimas necessárias para o detalhamento dos glicosaminoglicanos (GAGs).
a MPS é caracterizada por manifestações clínicas progressivas e sistémicas. Apesar de sua bioquímica e genética heterogeneidade, diferentes tipos de compartilhar características clínicas em combinações variadas, incluindo a articulação e displasia esquelética com rigidez (exceto MPS IV, onde há flacidez) e a dor, grosseiro características faciais, opacificação da córnea, inguinal ou abdominal, hérnias recorrentes do trato respiratório superior, infecções, doença de válvula cardíaca, síndrome do túnel do carpo, e a variável de envolvimento neurológico. Estas características aparecem geralmente nos primeiros meses de vida para as formas graves e na primeira infância para as formas mais atenuadas, mas são frequentemente subestimadas e geralmente tidas em conta apenas quando claramente evidentes.
a raridade destes distúrbios e a variabilidade na apresentação clínica conduzem frequentemente a um atraso de diagnóstico; este pode variar de meses, para as formas graves, a anos, para as atenuadas (ver Rigoldi et al. neste suplemento). No entanto, devido à rápida progressão e necessidade urgente de intervenção nas apresentações severas, mesmo meses de atraso no diagnóstico pode produzir resultados catastróficos para a saúde futura do paciente.
o nosso objectivo é sublinhar nesta revisão os sinais muito precoces das formas graves que devem alertar o pediatra sobre a sua primeira aparição.
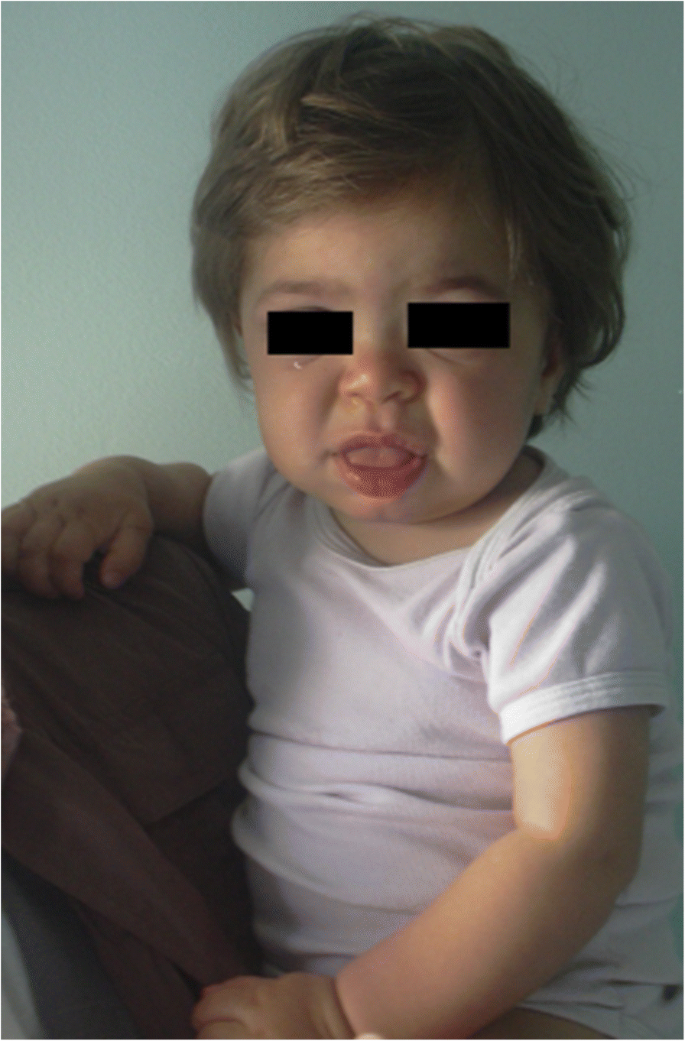
que sinais / sintomas podemos esperar em crianças com formas graves de MPS?Os sintomas e sinais sugestivos de diferentes formas de MPS são relatados na Tabela 1 de acordo com a idade. Nos primeiros 6 meses de vida, hérnias inguinais, infecções respiratórias anormalmente frequentes, otite e organomegalia podem ser observadas em doentes com MPS, particularmente em MPS i (síndrome de Hurler), que tem o início mais precoce grave. Além disso, de 6 a 12 meses, estas crianças muitas vezes desenvolvem um Gibbo (cifose toraco-lombar), sopro cardíaco, hérnia umbilical, hipotonia leve, e atraso de crescimento . Eles também podem ter a aparência facial típica (Fig. 1). MPS II, VI e VII podem apresentar um fenótipo similar. Os doentes com MPS IV apresentam um fenótipo esquelético precoce com o aparecimento de displasia da anca nos primeiros meses de vida e peito de pombo (pectus carinatum) nos primeiros 12-18 meses. Crianças MPS II presentes com um envolvimento semelhante, mas com início posterior. No segundo ano de vida, os bebês da síndrome MPS I Hurler desenvolvem atraso cognitivo, enquanto um paciente MPS II pode ser identificado apenas por causa do crescimento excessivo, infecções frequentes das vias aéreas e múltiplas cirurgias ; as características faciais típicas aparecem apenas mais tarde. Alguns doentes com MPS II também desenvolvem hiperactividade entre 18 e 24 meses. No terceiro ano de vida, hiperatividade e atraso cognitivo são facilmente reconhecíveis na MPS II e MPS III.
Tabela 1 Idade de aparecimento dos principais sinais e sintomas em diferentes mucopolysaccharidosis (MPS) tipos de
Fig. 1
MPS IH em um paciente de 1 ano. Faces grosseiras: ponte nasal plana, macroglossia, Boss frontais
em resumo, as manifestações esqueléticas são típicas e por vezes precoce. Faces grosseiras e atraso cognitivo não são sinais proeminentes precoces.
quais são as diferentes apresentações dos vários tipos de MPS?
MPS com envolvimento somático e cognitivo
mucopolissacaridose tipo I (MPS i; OMIM #252800) (figos. 1, 2, 3, 4 e 5) é causada por uma deficiência da hidrolase lisossómica α-l-iduronidase, o que leva à acumulação multissistemas de duas GAGs: sulfato de dermatano (DS) e sulfato de heparina (HS) . Com base na sua gravidade, a MPS I é tradicionalmente dividida em três fenótipos clínicos; no entanto, estas formas representam um continuum de gravidade da doença. A síndrome de Hurler, a forma mais grave, apresenta-se tipicamente durante o primeiro ano de vida. Crianças afetadas rapidamente desenvolvem disfunção cognitiva significativa e doença somática em vários sistemas de órgãos, levando à morte na primeira década na ausência de tratamento. As formas atenuadas de MPS i, conhecidas como síndrome de Hurler–Scheie e síndrome de Scheie, respectivamente, são caracterizadas pelo aparecimento posterior de sintomas, maior expectativa de vida e envolvimento leve ou não do sistema nervoso central (SNC). A herança é autossómica recessiva e, em pelo menos 50% dos casos, é possível prever fenótipos com base no genótipo, enquanto nos restantes 50% são detectadas novas mutações . A prevalência é de aproximadamente 1 em cada 100 mil nascimentos vivos .
Fig. 2
Polícia Militar. um Gibbus num paciente de 9 meses. raio-x da coluna vertebral num paciente de 3 anos. c imagem de ressonância magnética da coluna vertebral ponderada pelo T2, com cifose significativa e estenose do canal vertebral
Fig. 3
Polícia Militar. Raio-X prova de disostose multiplex. um espessamento das costelas num doente de 3 anos de idade e displasia da anca B noutro doente de 18 meses de idade
Fig. 4
MPS IH: uma mão de garra em um paciente de 18 meses de idade; b mão de raio-X.
Fig. 5
Polícia Militar. um espessamento do osso cortical do crânio e conformação anormal “em forma de J” da sella turcica. Dilatação dos espaços periventriculares em FLAIR b e imagens de ressonância magnética ponderadas c T-2 de um doente com 17 meses de idade
mucopolissacaridose tipo II (MPS II; OMIM # 309900) (Fig. 6), também conhecido como síndrome de Hunter, é o resultado da deficiência da enzima iduronato-2-sulfatase (I2S) com a consequente acumulação GAG de DS e HS . A prevalência é de cerca de 1 em 140,000–156,000 nascimentos vivos do sexo masculino . Notavelmente, este é o único X-linked MPS, e portadores femininos são geralmente assintomáticos; no entanto, eles podem ser excepcionalmente afetados devido a anormalidades do cromossomo X, homozigosidade, ou inactivação skewed-X -. A expressão fenotípica também abrange um amplo espectro de gravidade clínica. Em casos graves, coexistem o envolvimento neurológico progressivo e a doença somática, e a morte geralmente ocorre na segunda década de vida. Outros pacientes com disfunção neurológica mínima ou nenhuma pode apresentar um envolvimento somático grave enquanto, no extremo oposto do espectro, há pacientes com manifestações somáticas mínimas e inteligência normal que sobrevivem até a idade adulta . Sinais e sintomas precoces são compartilhados pelas formas graves de MPS I e II.
Fig. 6
MPS II. Um doente de 3 anos com apenas características faciais ligeiras
casos esporádicos de hidropis fetalis são descritos em MPS I, embora, em geral, estas crianças parecem normais no nascimento. Os primeiros sinais de MPS I geralmente aparecem durante os primeiros meses de vida, enquanto aqueles em MPS II geralmente ocorrem um pouco mais tarde . Kiely et al. em um estudo retrospectivo em 55 Hurler pacientes, observou-se que os sintomas respiratórios, dificuldades de amamentação, hérnia inguinal, e otite média tinha aparecido em uma idade mediana ≤ 6 meses, e a cifose, malformações cardíacas, aumento da circunferência da cabeça, hipotonia, organomegalia, opacificação da córnea, conjunto de restrição, e hérnia umbilical apareceu entre 6 e 12 meses. Hérnia Umbilical é uma das mais antigas características de apresentação em MPS I e II, e hérnias inguinais são relatadas em aproximadamente 60% dos pacientes .
outra pista inicial comum para MPS i e II pode ser infecções recorrentes nas vias aéreas superiores com aumento das secreções. A otite frequente, a rinite crónica recorrente e a descarga nasal persistente sem infecção óbvia não são específicas, mas podem ajudar a reforçar a suspeita diagnóstica se associada a sinais mais específicos. O armazenamento de GAG dentro da oro-faringe leva ao aumento das amígdalas e adenóides, e contribui para complicações nas vias aéreas superiores. Outra descoberta frequente é a respiração ruidosa, particularmente à noite, associada nas fases posteriores à apneia obstrutiva do sono. Como consequência de frequentes infecções no ouvido médio e disostose dos ossículos do ouvido médio, a perda auditiva é frequente, com défices condutivos e sensorineurais . Tracheobronchomalacia é comumente observada e pode levar a obstrução aguda das vias aéreas ou colapso, sendo esta uma das principais causas de morte nos anos seguintes .
adenoidectomia, tonsilectomia e timpanostomia, juntamente com reparação de hérnia, são as intervenções cirúrgicas mais frequentes e precoces para pacientes MPS I e II, frequentemente realizadas antes de conhecer o diagnóstico (em mais de 50% dos casos) .
a diarreia recorrente é bastante comum nos estadios iniciais da MPS i, e pode também estar presente em doentes com envolvimento neurológico da MPS II, embora seja pouco frequente nos doentes ligeiramente afectados .
deformidade Gibbus (cifose dorso-lombar) (Fig. 2) torna-se clinicamente aparente com uma idade média de 8 anos.6 meses a 1 ano no MPS i, representando um marco muito específico e precoce dos MPS em geral. Os corpos vertebrais parecem achatados e bicados, levando potencialmente a complicações posteriores como entalamento do nervo espinhal, lesão espinhal aguda e instabilidade atlanto-occipital; assim, deve-se ter especial cuidado para evitar a deslocação da articulação atlanto-axial durante a anestesia geral e cirurgia. Após 1 ano de idade, as contraturas articulares e genu valgum representam outros sinais freqüentes de disfunção articular e displasia esquelética progressiva, que são facilmente detectáveis em mais de 2 anos de idade em todos os bebês afetados pela MPS. Todas estas manifestações esqueléticas juntas, típicas da MPS, são chamadas de disostose multiplex. Espessamento das costelas pode ser visto em radiografias (Fig. 3a), os ossos longos são curtos com veios largos, e os joelhos são propensos a deformidades valgus e varus. Disostose falangeal e espessamento sinovial levam a uma deformação característica da mão da garra(Fig. 4), que é outra característica típica que pode levantar suspeitas da doença. A displasia da anca é frequentemente a razão pela qual estas crianças podem ser vistas pela primeira vez por especialistas ortopédicos. Tipicamente, a pélvis é mal formada, as cabeças femorais são pequenas, e coxa valga é comum, com deformidade progressiva e debilitante do quadril (Fig. 3b) .
existe uma diferença notável entre MPS I e MPS II; em MPS i, o crescimento do esqueleto desacelera aos 3 anos de idade , enquanto que os pacientes com MPS II apresentam crescimento excessivo nos primeiros 5-6 anos de idade, retardando o crescimento a partir daí .
Engrossamento das características faciais (nariz largo com dilatou as narinas, destaque supraorbital cristas, grande arredondado bochechas, lábios grossos, ampliada salientes língua), causada por armazenamento de GAGs nos tecidos moles da região orofacial e ossos da face se torna evidente dentro dos primeiros 2 anos, em uma idade mediana de 1,1 anos para a doença de Hurler e entre os 18 meses e os 4 anos na forma grave de Caçador de doença , apesar de sutis alterações fenotípicas podem ser detectados mais cedo (desde a segunda metade do primeiro ano), por pediatras com experiência específica (Fig. 1). A macrocefalia torna-se progressivamente mais evidente, e a scafocefalia é comum (Fig. 5), mas a microcefalia também é possível em pacientes com Hurler . O hirsutismo Facial e corporal são sempre evidentes aos 24 meses de idade . A pele pode ser espessada e inelástica. Notavelmente, alguns pacientes MPS II desenvolvem uma lesão cutânea distintiva, que é descrita como pápulas de cor branca-de-marfim (pebble-like) na parte superior das costas e nos lados superiores dos braços, patognomônica da síndrome de Hunter .
cardiomiopatia e espessamento progressivo e endurecimento dos folhetos da válvula em doentes com MPS I podem levar a regurgitação mitral e, menos frequentemente, aórtica . Um sopro pode ser detectado nos primeiros meses de vida e às vezes tem sido relatado como o primeiro sinal da doença . O envolvimento cardíaco também está presente em quase todos os pacientes com MPS II, mas isso começa com cerca de 5 anos de idade . A doença da válvula pode levar a hipertrofia ventricular direita e esquerda e insuficiência cardíaca.A protuberância do abdómen causada por hepatosplenomegalia progressiva é facilmente evidente após os 6 meses de idade, embora a conservação de GAGs no fígado e baço não conduza a disfunção orgânica .
a turvação da córnea ocorre em quase todos os indivíduos com MPS I com menos de 15 meses de idade , causando potencialmente deficiência visual grave. Pelo contrário , a turvação da córnea não é uma característica típica da síndrome de Hunter, mas o exame da lâmpada de fenda pode revelar lesões da córnea discretas que não afetam a visão. Disfunção da retina pode ser detectada, enquanto glaucoma não é um achado comum.
o desenvolvimento psicomotor precoce é geralmente detectado como normal, mas no MPS I atraso de desenvolvimento é geralmente óbvio pela idade de 18 meses, com um declínio mental progressivo levando a uma grave deficiência intelectual. As competências linguísticas são muito limitadas nestas crianças. Um atraso global dos marcos do desenvolvimento em MPS II torna-se evidente a partir de 2 anos em diante . No entanto, a principal característica neurológica da doença grave do Caçador é representada por distúrbios comportamentais marcados, tais como hiperatividade, obstinação e agressividade, que normalmente não são observados naqueles com o fenótipo atenuado .
mucopolissacaridose tipo VII (MPS VII; OMIM #253220), também conhecido como síndrome de Sly, é um MPS ultra-raro caracterizado pela atividade deficiente de β-glucuronidase, com armazenamento lisossomal de sulfato de condroitina (CS), DS e HS, levando a disfunção celular e de órgãos. O primeiro paciente foi descrito por Sly et al. em 1973 e um total de 145 pacientes foram relatados na literatura até agora.
a apresentação clínica e progressão da doença de MPS VII abrange um amplo espectro de gravidade, desde manifestações precoces, graves, multissistemas e morte nos primeiros meses, até um fenótipo mais leve com início posterior, inteligência normal ou quase normal, e sobrevivência mais longa .
Fenótipo características dos pacientes com MPS VII assemelham aos de MPS I e MPS II (baixa estatura, displasia esquelética, macrocefalia, hipertrofia gengival, recorrentes infecções de ouvido, hepatoesplenomegalia, hérnias, com envolvimento cardíaco, diminuição da função pulmonar e disfunção cognitiva). Uma proporção inesperadamente elevada de pacientes descritos (41%) tem história de hidrops fetalis não imunes neonatais (NIHF) . Apesar do início pré-natal da doença nestes pacientes, 13 em 23 sobreviveram à infância com um curso leve a intermediário no final da adolescência. Assim, a presença de NIHF não prevê, por si só, a possível gravidade do curso clínico se o paciente sobreviver à infância. Outro sinal inicial é macrocrania, enquanto o envolvimento da válvula cardíaca e infecções repetidas do trato respiratório superior e inferior aparecem nos primeiros 2 anos de vida. Atraso mental moderado e perda auditiva progressiva com deficiência da fala também são evidentes com o tempo.
em resumo, as formas graves de MPS I têm um início muito precoce do multissistema (dentro de 6 meses de vida).; MPS II é semelhante, mas com um início posterior, e MPS VII tem um início pré-natal com frequente NIHF e envolvimento grave precoce do trato respiratório superior e inferior.
MPS com envolvimento neurológico e cognitivo na sua maioria
Mucopolissacaridoses tipo III (MPS III, também conhecido como síndrome de Sanfilippo; Fig. 7) tem uma apresentação neurológica prevalente. MPS III é um distúrbio de armazenamento lisossômico causado pela deficiência de uma das quatro enzimas envolvidas no catabolismo do HS. Quatro subtipos diferentes são conhecidos—MPS III tipo-A (OMIM #252900), tipo B (OMIM #252920), tipo C (OMIM #252930), e do tipo D (OMIM #252940)—cada devidas a diferentes deficiência enzimática: tipo A, sulfamidase/SGSH gene; tipo B, alfa-N-acetylglucosaminidase/NAGLU gene; tipo C, alfa-glucosaminide N-acetyltransferase/HGSNAT gene; tipo D, N-acetilglucosamina-6-solfato sulfatasi/GNS gene). Todos os quatro tipos (A A D) têm herança autossômica recessiva. MPS III é o mais frequente dos MPS com uma prevalência estimada entre 0,3 e 4.1 casos para cada 100.000 recém-nascidos, dependendo do subtipo e raça .
Fig. 7
MPS IIIA. O mesmo paciente com 2,5 e 5 anos de idade. Note-se a expressão facial diferente, mostrando progressão da disfunção neurológica
os doentes com síndrome de Sanfilippo apresentam uma diminuição cognitiva progressiva no segundo e terceiro anos de vida. Em contraste com a maioria dos Deputados, características somáticas são relativamente menos evidentes, e o envolvimento do SNC é conspícuo. O atraso no desenvolvimento da fala é geralmente o primeiro sinal notado pelos pais, mas as principais preocupações podem ser problemas comportamentais (hiperactividade, comportamento ansioso e agressivo, distúrbios do sono), seguidos de declínio mental progressivo . Estes sintomas podem causar mal diagnóstico como distúrbios comportamentais, transtorno do déficit de atenção hiperatividade (TDAH), e distúrbios do espectro do autismo . Pode também estar presente epilepsia.
distúrbios do sono, cuja incidência é relatada até 80-90%, são uma característica comum. Eles consistem em dificuldades em adormecer e despertar noturno frequente, com inversão completa do ritmo dia–noite em alguns pacientes.
todas estas manifestações são muito difíceis de gerir e têm um enorme impacto psicológico na qualidade de vida de toda a família.
em geral, embora o fenótipo possa ser muito semelhante nos quatro subtipos, o curso clínico em Sanfilippo B E C parece ser caracterizado por um fenótipo menos grave .
os sintomas não neurológicos são geralmente menos pronunciados em MPS III do que nos outros MPS. Infecções recorrentes do ouvido, nariz e garganta são observadas em uma idade mais jovem. Otite recorrente, defeitos no ouvido médio e anomalias no ouvido interno podem causar surdez. Um excesso de secreções espessas e alterações anatômicas podem produzir obstrução das vias aéreas. Nos primeiros anos de vida, a diarreia é frequentemente notificada, enquanto que a obstipação é mais comum em doentes mais velhos.
o exame físico pode mostrar características faciais grosseiras, embora estas sejam menos pronunciadas. Os pacientes têm macrocefalia, sobrancelhas largas com inflamação medial e sinofrias, o cabelo é geralmente seco e grosso, e hipertricose é comum. Em doentes mais jovens, observa-se frequentemente hepatomegalia ligeira e hérnias umbilical e inguinal. As contraturas raras e muitas vezes suaves são encontradas principalmente nos cotovelos. O crescimento é afectado em aproximadamente metade dos doentes com mais de 12 anos .
em resumo, a doença de MPS III ou Sanfilippo tem um envolvimento neurológico proeminente com sinais somáticos muito ligeiros. Crianças de 2 a 3 anos de idade que desenvolvem hiperactividade, TDAH e comportamento agressivo podem ser suspeitas de terem MPS III.
MPS com envolvimento somático prevalente
MPS IV e MPS VI presentes apenas com envolvimento somático. Estas são condições progressivas que poupam capacidade intelectual e afetam principalmente o esqueleto (MPS IVA), ou todos os órgãos/sistemas do corpo (MPS VI). A herança é autossómica recessiva. A taxa de agravamento dos sintomas varia entre os indivíduos afetados.
mucopolissacaridose IV (MPS IV, também conhecido como síndrome de Morquio; Fig. 8) apresenta-se como duas geneticamente distintas transtornos, cada um com um diferente deficiência enzimática: MPS IVA (Morquio Uma síndrome; OMIM #253000) devido à deficiência de N-acetylgalactosamine-6-sulfatase (GALNS gene), resultando no acúmulo de keratan-sulfato (KS) e CS ; e MPS IVB (Morquio B síndrome; OMIM #253010) devido à deficiência de beta-galactosidase a atividade, levando a urina KS e oligossacarídeo excreção como em GM1 pacientes. MPS IVB é de fato alélico para as várias formas de gangliosidose GM1, mas não tem a deterioração psicomotora vista na gangliosidose GM1 .
Fig. 8
MPS IVA. um de 4 anos, o paciente com b antero-posterior e lateral c de raios-x mostrando a “remar” em forma de costelas e achatado e arredondado vértebras com um típico “anterior beaking” aspecto
Pacientes com grave da MPS IVA, sem detectável a atividade da enzima revelar seus primeiros sintomas geralmente durante o primeiro ano de vida, embora eles raramente são diagnosticados antes de 4-5 anos de idade. A informação sobre a história natural dos pacientes Morquio A é derivada principalmente de duas grandes coleções de dados . A taxa de crescimento reduzida começa com aproximadamente 18 meses de idade e o crescimento vai parar com aproximadamente 7 ou 8 anos de idade. Em contraste com os outros MPS, as articulações Morquio A são geralmente laxantes e muito flexíveis (hipermobile), mas algumas articulações (geralmente os quadris e ombros) podem ter uma faixa restrita de movimento. Os doentes com tal envolvimento esquelético podem receber um diagnóstico ou ser submetidos a avaliação de displasia espondiloepifisal, pseudoachondroplasia, displasia epifiseal múltipla ou doença bilateral de Legg-Calvé-Perthes .
uma característica constante é hipoplasia odontóide; assim, o deslocamento da articulação atlanto-occipital pode ocorrer causando compressão e danos à bula e medula espinhal, resultando em paralisia ou até mesmo morte se a estabilização cervical não é realizada precocemente.
os doentes com MPS IVB têm uma manifestação clínica muito semelhante, mas com uma estatura curta menos proeminente. Eles têm características faciais levemente grosseiras, perda auditiva, opacidades da córnea, doença cardíaca valvular, e infecções freqüentes do trato respiratório superior. Podem também apresentar hérnia inguinal e hepatomegalia ligeira. Características displásicas esqueléticas incluem platipondilia, hipoplasia odontóide com possível subluxação cervical, escoliose kypho, coxa valga, e asas ilíacas constritas .
mucopolissacaridose VI (MPS VI, também conhecido como síndrome de Maroteaux-Lamy; OMIM # 253200; Fig. 9) é devido a mutações patogénicas no gene da arilsulfatase B (ARSB) localizado no cromossoma 5. Consequentemente, a actividade reduzida ou ausente da enzima arilsulfatase B (ASB) (n-acetilgalactosamina 4-sulfatase) prejudica a degradação do DS que determina a sua acumulação celular .
Fig. 9
MPS VI. Um 2-year-old boy, com evidente grossas e rosto esquelético disostose
Pacientes com detectaveis, a atividade da enzima têm a forma grave da MPS VI e apresentar sintomas no início da infância. Seu fenótipo somático é semelhante à síndrome de Hurler. Na maioria dos casos, aos 2 ou 3 anos de idade, lesões ósseas graves e progressivas, chamadas disostose multiplex, torna-se evidente. Este displasia esquelética inclui displasia da anca com displásicas cabeça femoral, anormal de corpos vertebrais, irregular clavículas, hipoplasia distal ulna e o raio, e displásicas e metacarpo curto ossos; do carpo e do tarso ossos são hipoplasia e irregular perfil. A velocidade de crescimento geralmente diminui após o primeiro ano de vida, com uma altura final geralmente inferior a 120 cm. Como em outros MPS, um sinal inicial é características faciais grosseiras. Além disso, os doentes têm um abdómen saliente típico, hepatomegalia, hérnia umbilical e/ou inguinal, e envolvimento cardíaco no início da infância. Embora não esteja presente nenhuma deficiência intelectual primária, a hidrocefalia pode ser vista em alguns pacientes com conseqüente hipertensão intracraniana e papiledema. Pectus carinatum, articulações rígidas e contraídas, escoliose e cifose e compressão da espinal medula cervical pioram com o tempo .
em resumo, MPS IV e VI não mostram envolvimento do SNC. MPS IV é um MPS único em Mostrar laxidade nas articulações, enquanto o aparecimento grave de MPS VI é semelhante ao observado em MPS I.