Autosomal Recessive Congenital Ichthyosis / Actas Dermo-Sifiliogr Exceptionficas
Inledning
den senaste konsensusklassificeringen av ichthyosis skiljer mellan 2 huvudformer: de icke-syndromiska formerna, som endast uppvisar hudmanifestationer, och de syndromiska formerna, som också uppvisar manifestationer i andra organ (Tabell 1).1 bland de icke-syndromiska formerna identifieras 4 grupper: vanliga ichthyoser, autosomala recessiva medfödda ichthyoser (ARCIs), keratinopatiska ichthyoser och andra mindre vanliga ichthyoser.Traditionellt var gruppen av ARCIs uppdelad i 2 störningar, lamellär ichthyosis (LI) och medfödd ichthyosiform erythroderma (CIE). I den nya klassificeringen tillsattes Harlekin-iktyos (HI) till denna grupp1 eftersom inaktiverande mutationer i ABCA12-genen har identifierats som ansvariga för denna störning,2,3 medan nonsensmutationer i samma gen kan ge upphov till LI4-eller CIE5,6-fenotypen. Andra mindre vanliga varianter som ingår i gruppen ARCIs är självläkande kollodion baby (SHCB), acral SHCB och baddräktiktyos.7-9
Konsensusklassificering baserad på de kliniska egenskaperna hos Ichthyosis1.
| icke-Romiska former | Syndromiska former |
| vanlig Iktyosichthyosis vulgarisrecessiv x-länkad iktyos (nonsyndromic )ajimajor formerharlekin iktyosislamellär iktyosmedfödd iktyosiform erythrodermaMinor formersjälvläkande kollodion babyakral självläkande kollodion babybaddräkt iktyosiskeratinopatisk Iktyosstora formerepidermolytisk iktyosytlig epidermolytisk iktyosisminor formerannulär epidermolytisk iktyosiskurth-Macklin Ichthyosisautosomal recessiv epidermolytic ichthyosisEpidermolytic nevusandra Formerloricrin keratodermaErythrokeratodermia vararabilispeeling hudsyndrommedfödd retikulär ichthyosiform erythrodermaKLICK syndrom | syndromisk X-länkad Ichthyosrecessiv x-länkad ichthyosis (syndromisk)ichthyosis follicularis, alopeci och fotofobi (IFAP) syndromconradi-Scammann-happle syndrom (chondrodysplasia punctata typ 2)autosomala iktyosishudstörningarneterton Syndromiktyos-hypotrichos syndromiktyos-skleroserande kolangitsyndromtrichotystrofyneurologiska störningarj syndromeRefsum diseaseMEDNIK syndromeFatal disease courseGaucher disease, type 2Multiple sulfatase deficiencyCEDNIK syndromeARC syndromeOther associated signsKID syndromeChanarin-Dorfman syndromeIchthyosis prematurity syndrome |
Abbreviations: ARC, arthrogryposis–renal dysfunction–cholestasis; ARCI, autosomal recessive congenital ichthyosis; CEDNIK, cerebral dysgenesis, neuropathy, ichthyosis, and palmoplantar keratoderma; KID, keratitis ichthyosis deafness; KLICK, keratosis linearis with ichthyosis congenital and sclerosing keratoderma; MEDNIK, mental retardation, enteropati, dövhet, perifer neuropati, iktyos, keratoderma.
endast begränsade data finns tillgängliga om Arcis epidemiologi. I Förenta staterna har en prevalens vid födseln av 1 per 100 000 befolkning för LI och av 1 per 200 000 befolkning för CIE uppskattats. Andra studier har rapporterat en kombinerad prevalens för LI och CIE på 1 per 200 000 till 300 000 populationer.10,11 i vissa länder som Norge är den uppskattade prevalensen större (1 per 91 000) på grund av grundmutationer.12 upptäckten av 1 eller flera återkommande mutationer i en population kan bero på att mutationen inträffade vid en given punkt i historien och sedan överfördes från generation till generation (grundmutation) eller för att regionen i genomet där mutationen finns har en DNA-sekvens mottaglig för mutation (mutation hotspot). I Spanien är den uppskattade prevalensen av ARCI 1 per 138 000 i den allmänna befolkningen och 1 per 61 700 bland barn under 10 år.13 i vissa regioner i Spanien kan förekomsten bli ännu högre. På den galiciska kusten rapporterades till exempel en prevalens på 1 per 33 000, också på grund av en grundareffekt.14
lamellär iktyos och medfödda Iktyosiforma Erythrodermakliniska egenskaper
även om man ursprungligen trodde att LI och CIE var olika enheter, har det rapporterats om patienter med mellanliggande kliniska manifestationer och båda tillstånden kan orsakas av mutationer i samma gen.15,16 dessutom kan patienter med samma mutation, även inom samma familj, utveckla olika fenotyper.12,15
de flesta patienter föds inneslutna i ett kollodionmembran som gradvis försvinner under de första veckorna av livet och ersätts av den definitiva fenotypen (Fig. 1A). Hypohidros, svår värmeintolerans och nageldystrofi observeras ofta i både LI och CIE.17-19 patienter med LI har vanligtvis allvarligare kliniska manifestationer än de med CIE. De har stora plattliknande skalor, ofta av mörk färg, som täcker hela kroppsytan. Erytroderma är antingen frånvarande eller minimal. Sådana patienter har vanligtvis ectropion och ibland eclabium, hypoplasi av LED-och näsbrosk, ärrbildning alopeci, särskilt vid kanten av hårbotten och palmoplantar keratoderma (Fig. 1B och C). CIE kännetecknas av närvaron av erytroderma och fin vitaktig skalning (Fig. 2). Vissa patienter har märkt erytem och generaliserad skalning. Vågen kan vara stora och mörka, särskilt på benens extensorytor. I mindre allvarliga fall är erytem milt och skalningen är bra.

kliniska egenskaper hos lamellär iktyos. En, brunaktig lamellär desquamation. B, märkt plantar hyperkeratos. C, ärrbildning alopeci i hårbotten.
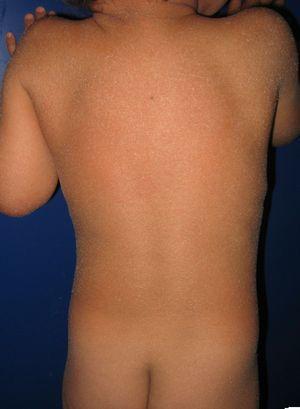
Patient med medfödd iktyosiform erytroderma och mutationer i ALOXE3-genen. Mild erytem och generaliserad vitaktig furfuraceous desquamation kan ses.
Histopatologi
histopatologiska förändringar ger ingen diagnos. I LI observeras massiv ortokeratotisk hyperkeratos, vanligtvis med två gånger förlängningen som i CIE. Epidermis är akantotisk och får ibland ett psoriasisliknande utseende. Cellproliferationshastigheten är normal eller något förhöjd.17-19 patienter med CIE har mindre markerad hyperkeratos, med fokal eller omfattande parakeratos, ett normalt eller förtjockat granulärt skikt och mer uttalad akantos. Den epidermala omsättningen ökar.17-19
ultrastruktur
även om en nära korrelation mellan molekylära, kliniska och ultrastrukturella fynd hittills inte har hittats, kan elektronmikroskopi ändå vara användbar för att utesluta andra former av iktyos och för att styra genetiska analyser i vissa fall. Fyra typer av medfödd iktyos har beskrivits (Tabell 2).
ultrastrukturell klassificering av medfödda Ichthyoser.
| Typ | huvudfunktion | andra funktioner | mutationer | kliniska manifestationer |
| 1 | frånvaro av ultrastrukturella markörer av iktyos typ 2, 3 och 4 | lipiddroppar eller ringar i stratum corneum (mest frekventa)små keratohyalin granulesvesikulära eller lobulära membranbeläggningsgranuler | TGM1 (33.3%)ALOX12B (2 fall) | CIE |
| 2 | kolesterol klyftor i stratum corneum | frånvaro eller gallring av cornified envelopesmå keratohyalin granulesLipid droppar | TGM1 (89-100%) | LI |
| 3 | laminerade membranstrukturer i stratum granulosum och / eller stratum corneum. | onormal membranbeläggning granulerlipid dropparfoci av framträdande juxtanukleära vakuoler i det granulära skiktet | NIPAL4 (93%) | CIE (vanligast)LI |
| 4 | Trilamellära membranpaket som fyller vissa celler i stratum granulosum och/eller stratum corneum | onormala membranbeläggningsgranuler | FTAP4 | iktyos prematuritetssyndrom(100%) |
förkortningar: CIE, medfödd iktyosiform erytroderma; LI, lamellär iktyos.
medfödd iktyos typ 1
medfödd iktyos typ 1 kännetecknas av frånvaron av ultrastrukturella markörer för iktyos typ 2, 3 och 4. Därför ställs diagnosen vanligtvis bara när de andra typerna har uteslutits. Det vanligaste fyndet är närvaron av lipiddroppar eller ringar i stratum corneum (Fig. 3A).20 dessa lipiddroppar är inte en konstant egenskap eller specifik för denna speciella typ eftersom de inte är närvarande i alla fall,20 och de kan vara närvarande i andra typer av iktyos.21,22 kliniskt har de flesta patienter med manifestationer av CIE.12,20 en tredjedel av patienterna har mutationer i tgm1-genen.16 denna ultrastrukturella typ har också identifierats i samband med mutationer i ALOX12B-genen.23,24

Elektronmikroskopbilder. A, medfödd iktyos typ 1, som visar lipiddroppar i stratum corneum och frånvaro av ultrastrukturella markörer för de andra typerna av iktyos. B, medfödd iktyos typ 2, kännetecknad av närvaron av kolesterolklyftor (pil) i corneocyter.
medfödd iktyos typ 2
medfödd iktyos typ 2 kännetecknas av kolesterolklyftor i stratum corneum (Fig. 3B).21 sådana klyftor är ett konstant fynd i denna typ av iktyos och kan detekteras i olika biopsier hos samma patient; behandling med orala retinoider har ingen inverkan på dessa klyftor.12,25 Elektrontäta aggregat har också observerats på corneocyter hos vissa patienter med bristfällig Tgas 1-aktivitet.26-28 kliniskt har de flesta patienter allvarliga manifestationer av CIE.12 denna ultrastrukturella typ är starkt associerad med mutationer i tgm1-genen.12,16
medfödd iktyos typ 3
medfödd iktyos typ 3 kännetecknas av lamellära membranstrukturer i stratum granulosum och/eller stratum corneum. Dessa strukturer är anordnade i remsor runt ett tomt utrymme nära kärnan.22,29 – 31 de kliniska manifestationerna av denna typ skiljer sig från de andra; uppkomsten av iktyos är variabel, desquamation och erytem kan vara fläckig eller generaliserad, och särskilt böjningarna påverkas. Mutationer i nipal4-genen är ansvariga för 93% av ichthyoses typ 3.32
medfödd iktyos Typ 4
karaktäristiskt, vid medfödd iktyos typ 4, fylls vissa celler i stratum granulosum och stratum corneum med trilamellära membranpaket.33 dessa fynd är patognomiska för iktyos prematuritetssyndrom, ett tillstånd som för närvarande betraktas som en syndromisk form av iktyos.34,35
molekylära studier
i genetiska termer är ARCIs mycket heterogena. Tgm1-genen är associerad med de flesta fall, men mutationer i 5 andra gener (ALOX12B, ALOXE3, NIPAL4, CYP4F22 och ABCA12) har rapporterats. Fischer et al.36 studerade 520 familjer med ARCI och identifierade mutationer i minst 1 av dessa gener i 78% av fallen (TGM1 i 32%, NIPAL4 i 16%, ALOX12B i 12%, CYP4F22 i 8%, ALOXE3 i 5% och ABCA12 i 5%). I en annan studie av 250 patienter med ARCI av olika ursprung hade 38% tgm1-mutationer, 6, 8% hade ALOXE3-mutationer och 6, 8% hade ALOX12B-mutationer.37 i Galicien identifierade vi mutationer i generna TGM1, ALOX12B, ALOXE3, NIPAL4 och CYP4F22 i 75% av de studerade familjerna, men fördelningen av mutationer var annorlunda.14 tgm1-genen muterades i 68.7% av fallen medan ALOXE3-genen muterades på bara 1 patient. Vi upptäckte inte mutationer i någon av de andra 3-generna som studerades.
TGM1
tgm1-genen är belägen på kromosom 14q11.2 och har 15 exoner (GenBank NM-000359.2). Det kodar för Tgas 1-enzymet, vilket är ett av de 3 tgasenzymerna som finns i epidermis.38 detta enzym deltar i bildandet av det kornade höljet genom att katalysera kalciumberoende tvärbindning av flera proteiner såsom involucrin, loricrin och prolinrika proteiner.39,40 det katalyserar också bindning av ??- hydroxiceramider i det yttre skiktet av det kornade kuvertet med proteiner i det inre skiktet.41,42 hos patienter med tgm1-mutationer saknas det kornade kuvertet och Tgas 1-aktiviteten är reducerad eller obefintlig.43-47
sedan 1995, när denna gen identifierades som ansvarig för vissa fall av ARCI,har 48-50 mer än 110 mutationer rapporterats hos patienter med olika ursprung. Mutationer i TGM1 är den vanligaste orsaken till ARCI.36,37 denna mutation har hittats i 55% av fallen i USA och i 84% av fallen i Norge.12,51 den vanligaste mutationen är c.877-2a>G, som har hittats i 34% av de muterade allelerna som hittills rapporterats.52 den höga frekvensen av denna mutation i länder som USA och Norge beror på en grundareffekt.12,53 den näst vanligaste mutationen är p. Arg142His. Denna och liknande mutationer har rapporterats i länder som Egypten, Tyskland, Finland och USA, 15,49-51,54-56 och det verkar som om dessa är hotspot-mutationer.57 p. Arg307Trp-mutationen är frekvent i den japanska befolkningen.5 i Galicien, p. Arg760X, c.1223_1227delacaca och c.984 + 1g>a mutationer i TGM1 identifierades i 81,82% av familjerna med mutationer i denna gen, vilket tyder på en grundareffekt.14 bekräftelse av denna hypotes erhölls genom haplotypstudie (arbete som ännu inte publicerats).
tgm1-mutationer är ansvariga för de flesta fall av LI15,27,44,46,56,58-63 och för en liten andel fall av CIE.43,47,64,65 sådana mutationer kan också ge upphov till andra former av ARCI såsom SHCB, acral SHCB och baddräktiktyos.
många studier har försökt visa genotyp-fenotypföreningar mellan mutationer i TGM1 och ultrastrukturella eller kliniska fynd, men ingen signifikant korrelation har hittills observerats.15,16,53 i allmänhet påverkas patienter med mutationer i tgm1-genen mer allvarligt än de utan sådana mutationer. I en studie av 83 patienter med ARCI i Sverige och Estland var närvaron av ektropion och kollodionbarn associerad med tgm1-mutationer, medan en högre erytemfrekvens observerades hos patienter utan mutationer i denna gen.66 en annan studie visade att typen av skalning är den största skillnaden mellan bärare och icke-bärare av tgm1-mutationer, när man fann att alla patienter med mutationer i denna gen hade lamellskalning medan 80% av de utan tgm1-mutationer hade fin skalning.14 dessutom har man sett att trunkerande mutationer oftare förknippas med hypohidros och svettningsstörningar än missense-mutationer.51 i den nordamerikanska befolkningen förutsäger en modell baserad på förekomsten av vissa kliniska egenskaper att patienter som är födda som kollodionbarn och har okulära störningar och/eller alopeci är 4 gånger mer benägna att ha tgm1-mutationer.51
ALOXE3 och ALOX12B
ALOXE3-och ALOX12B-generna finns på kromosom 17p13.1. 67 de har en liknande struktur med 15 exoner som kodar för epidermal LOXs eLOX-3 och 12R-LOX.68,69 det faktum att de huvudsakligen uttrycks i epidermis suprabasala skikt stöder deras roll i avancerade faser av epidermal differentiering, med deltagande i behandlingen av lamellära kroppar.24,70 dessa enzymer verkar på intilliggande steg i hepoxilinvägen (Fig. 4). 12R-LOX omvandlar arakidonsyra till 12R-hydroxyeikosatetraensyra medan eLOX – 3 omvandlar denna produkt till en epoxialkoholisomer69,71 av hepoxilin A3-familjen.72 hepoxilin-produkten är instabil och hydrolyseras i celler till ett specifikt trihydroxiderivat (trioxilin). Även om den exakta rollen för produkterna i hepoxilinvägen inte är känd har det spekulerats i att de kan delta i bildandet av intercellulära lipider i stratum corneum eller fungera som signaler för att inducera keratinocytdifferentiering.
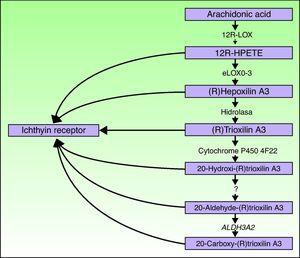
schematiskt diagram över hepoxilinvägen, som visar deltagandet av ALOXE3 -, ALOX12B -, NIPAL4-och CYP4F22-generna. Mutationer i dessa gener är ansvariga för vissa typer av ARCI. HPETE indikerar hydroperoxikosatetraensyra.
ALOX12B-och ALOXE3-generna identifierades först 2002.73, 74 sedan dess har mer än 30 mutationer i ALOX12B-genen 23,24,37,75-77 och cirka 10 i ALOXE3-genen 37,74,75 rapporterats. Dessa mutationer är ansvariga för 14% till 17% av ARCIs36,37 och 72.2% av SHCBs.23,78,79 orsakssambandet mellan dessa mutationer och fenotyp bekräftades genom att visa att den katalytiska aktiviteten hos epidermal LOX helt avskaffades hos patienter med dessa mutationer75,80 och genom att använda djurmodeller som reproducerade den iktyosiforma fenotypen som ses hos människor.81-83 båda generna är ansvariga för en liknande procentandel av ARCI-fall. Emellertid är utbudet av olika mutationer i ALOXE3-genen begränsad på grund av övervägande av 2 mutationer, p.Arg234X och p.Pro630Leu, som verkar motsvara hotspots.37,74,75
patienterna med mutationer i ALOXE3-och ALOX12B-generna visar vanligtvis en CIE-fenotyp.74,75,77 svårighetsgraden av skalning är mild eller måttlig, och skalorna har en vitaktig eller ljusbrun färg. Erytem kan också vara närvarande. Så många som 76% av patienterna är födda som kollodionbarn och 88% har svettningsstörningar.37 patienter med mutationer i ALOX12B-genen visar mer begränsad, vitaktig desquamation jämfört med bärare av mutationer i ALOXE3-genen. I dessa fall är skalorna brunaktiga och vidhäftande. Närvaron av erytem, palmoplantar hyperkeratos och accentuering av palmoplantarvecken är också associerade med ALOX12B-mutationer.37
Ichthyin/NIPAL4
nipal4-genen, även känd som ichthyingen, ligger på kromosom 5q33. Den har 6 exoner som kodar för ett protein med flera transmembrandomäner med okänd funktion.84 det har antagits att proteinprodukten deltar i samma metaboliska väg som LOX och kan fungera som en receptor för trioxiliner A3 och B3 eller för andra metaboliter av hepoxilin metabolisk väg.84 det skulle således vara inblandat i bildandet av lamellära kroppar eller i deras transport mot det extracellulära utrymmet.32 till stöd för detta är 2 observationer. För det första är mutationer i denna gen i 93% av fallen associerade med ett ultrastrukturellt mönster av medfödd iktyos typ 3, kännetecknad av abnormiteter i de lamellära kropparna och närvaron av långsträckta perinukleära membran i stratum granulosum.32 för det andra uttrycks NIPAL4 väsentligen i stratum granulosum av epidermis, där de lamellära kropparna är närvarande.85
sedan upptäckten av nipal4-genen 2004,84 har endast 9 mutationer rapporterats hos patienter från Medelhavsländerna (Algeriet, Turkiet och Syrien), 84 skandinaviska länder,32 Pakistan,85 Färöarna,32 och Sydamerika.84
det kliniska spektrumet hos patienter med mutationer i denna gen är bred, även bland medlemmar i samma familj. Mellan 3, 7% 32 och 60% 84 föds som kollodionbarn. När kollodionmembranet försvinner utvecklar de flesta patienter manifestationerna av CIE, med fina vita vågar på en erytematös bas i ansiktet och stammen och större, bruna skalor på nacken, skinkorna och benen.84 markerad xeros, generaliserade brunaktiga retikulära hyperkeratotiska plack som verkar accentuerade i hudvecken och ansiktsdyschromi kan vara närvarande.32,85 dessutom är palmoplantar keratoderma ett vanligt fynd tillsammans med tillfälliga fingerkontrakt och böjda fingernaglar. Vissa studier har rapporterat fynd som är mer typiska för LI.32,85 förekomsten av tecken och symtom på atopisk dermatit har rapporterats hos vissa patienter, även om mutationer i FLG-genen inte detekterades i något av dessa fall.85
CYP4F22
FLJ39501 eller cyp4f22-genen är belägen på kromosom 19p13.12. 86 den har 12 exons87 och kodar för en P450 cytokrom, familj 4, subfamilj F, polypeptid 2, homolog av leukotrien B4 – pankreas-hydroxylas (CYP4F2). Reaktionen som katalyseras av produkten av FLJ39501 i huden och substraten för den reaktionen kan härledas analogt med dess kända homologer CYP4F2 och CYP4F3.88 det har antagits att CYP4F2 och CYP4F3 deltar i hepoxilinvägen genom att katalysera omvandlingen av trioxilin A3 till 20-hydroxi-(R)trioxilin A387 och att slutprodukten av denna väg, 20-karboxi-trioxilin A3, kan ha en viktig biologisk reglerande effekt i huden.89
hittills har endast 8 mutationer av denna gen rapporterats i 12 konsanguineous familjer från Medelhavsländerna87 och i 1 familj av Israeliskt ursprung.62
i de familjer som rapporterats av lef Jacobvre et al., 87 de flesta patienter hade en cie-fenotyp vid födseln och detta utvecklades därefter till LI. patienter föddes vanligtvis med markant erytroderma, men utan något kollodionmembran. När de blev äldre utvecklade de generaliserad vitgrå skalning, som var mer markerad i periumbilical regionen, på skinkorna och på underdelen av kroppen. Hyperlinjäritet i handflatorna och fotsulorna och desquamation i hårbotten, vid tider av pityriasiform typ, var frekventa.87 i en annan familj föddes de 3 drabbade medlemmarna som kollidionbarn och utvecklade intensiv erytroderma, generaliserad desquamation och palmoplantar keratoderma.62
ABCA12
under 2003 rapporterades ABCA12-genen vara ansvarig för vissa fall av LI och kartlades till kromosom 2q34.4 Det bekräftades därefter att mutationer i denna gen också var ansvariga för HI.2, 3abca12 kodar 53 exoner och tillhör en familj av ABC-transportörer, som binder adenosintrifosfat samtidigt som de underlättar transporten av flera molekyler över cellmembranet.90 medlemmarna i ABCA-underfamiljen är alla inblandade i lipidtransport.91 bristfällig ABCA12-funktion orsakar lipidtransportstörningar i lamellära kroppar och leder därför till en minskning av intercellulära lipidnivåer i stratum corneum.3ultrastrukturella studier har visat att ABCA12 ligger i lamellära kroppar associerade med glykosylceramider.91abca12 mutationer har associerats med störningar i distribution och transport av glykosylceramider och med minskade nivåer av hydroxiceramider, en av huvudkomponenterna i lipidbarriären i de intercellulära utrymmena.3,6,92,93 den massiva hyperkeratos som förekommer hos dessa patienter kan vara ett kompensatoriskt svar på en bristfällig lipidbarriär.94 det kan också bero på bristen på desquamation av corneocyterna,93 som kan orsakas av defekter i transporten av vissa proteaser, såsom callicrein 5 och cathepsin D, som härrör från störningar i de lamellära kropparna.95 Murina modeller och in vitro-studier tyder på att ABCA12-mutationer också har en effekt på epidermal differentiering.95-97
hittills har mer än 50 mutationer rapporterats i ABCA12-genen hos patienter med ARCI från Afrika, Europa, Pakistan och Japan. De vanligaste mutationerna är P.Val244SerfsTer28,2, 98,99 identifierade i pakistanska och indiska populationer, och p.Asn1380Ser, 4 identifierade i afrikanska familjer. I båda fallen kan dessa vara grundande mutationer.
omfattningen av abca12-mutationerna är relaterad till fenotyp, med mutationer associerade med fullständig funktionsförlust som leder till HI-fenotypen.2,3,98-102 däremot i LI och CIE är de flesta mutationer missense och har en mindre allvarlig effekt på proteinfunktionen.4-6, 103 mutationerna som ligger bakom LI-fenotypen verkar koncentreras i den första adenosintrifosfatbindande kassettregionen.4 kliniskt har patienter med CIE och mutationer i ABCA12-genen medelstora skalor som är något större än de som vanligtvis observeras hos patienter med denna fenotyp.
Harlekin iktyos
hej eller harlekinfoster är en allvarlig och vanligtvis dödlig form av iktyos. Barnen är vanligtvis för tidiga med omfattande glänsande hyperkeratotiska plack, åtskilda av djupa sprickor, som täcker hela integumentet och bildar geometriska mönster som påminner om kläder som bärs av harlekiner, vilket ger tillståndet sitt namn. Hudtäthet leder till markant eversion av ögonlocken och läpparna, rudimentär utveckling av LED-och näsbrosk och ibland mikrocefali. Barnen har sällan ögonfransar eller ögonbryn, även om håret i hårbotten kan bevaras. Händer och fötter är svullna och edematösa och ofta täckta av ett handskliknande lager. De kan ha fingerkontrakt.
för sådana patienter är risken att dö under neonatalperioden mycket hög.104 lungventilation äventyras; transepidermal vattenförlust leder till uttorkning, vattenkraft obalans och termisk instabilitet; och risken för infektioner ökar. Ansiktstäthet och eclabium hindrar sugning och därför utfodring, med motsvarande försämring av uttorkning. Nyfödda med detta tillstånd levde sällan längre några veckor. Under de senaste åren har dock chanserna för långvarig överlevnad ökat särskilt, huvudsakligen på grund av administrering av systemiska retinoider och framsteg inom intensiv neonatalvård.105 i en ny studie överlevde 83% av patienterna som behandlades med orala retinoider jämfört med 24% av obehandlade patienter. De flesta av dödsfallen inträffade under de första 3 dagarna av livet, men behandlingen startades inte förrän efter detta hos många av de överlevande.104 detta tyder på att många av dessa tidiga dödsfall skulle ha inträffat oavsett retinoidbehandling.
barnen som överlever den nyfödda perioden utvecklar i allmänhet svår CIE.106 arten och placeringen av mutationer i ABCA12-genen och omfattningen av transportörfunktionsförlust kan bestämma prognosen.3,92,107 patienter som bevarar en viss grad av proteinaktivitet, om än minimal, kan ha en bättre chans att överleva. Bärare av homozygota mutationer har en högre dödlighet.104
den huvudsakliga histologiska egenskapen hos HI är närvaron av ett extremt tjockt och kompakt orthokeratotiskt stratum corneum. Hårsäckarna och svettkanalerna har framträdande hyperkeratotiska pluggar107, 108 och har onormala eller frånvarande lamellära kroppar, lipidinneslutningar eller rester av organeller eller kärnor i corneocyterna och frånvaro av intercellulära lipider i ultrastrukturell studie.108,109 hårsäckarna visar en markant koncentration av keratotiskt material, vilket är en diagnostisk egenskap hos HI som används för prenatal diagnos.
hittills är detekteringshastigheten för mutationer i ABCA12-genen hos patienter med HI nära 100%, och så verkar detta vara ett genetiskt homogent tillstånd.
Collodion Baby och självläkande Collodion Baby
Collodion babies föds vanligtvis för tidigt och perinatal sjuklighet och dödlighet ökar. Vid födseln täcks den nyfödda av ett glänsande undervisat transparent membran som påminner om cellofanförpackning (Fig. 5). Barnen har ectropion, eclabium och hypoplasi i näs-och ledbrosket. Sugande och lungventilation kan hindras110 och transepidermal förlust av vatten och risken för infektioner ökar.110,111

Kollodionbarn som därefter utvecklades till en lamellär iktyosfenotyp.
Collodion baby är den vanliga presentationen för HI och CIE. Autosomal dominant LI, 112, 113 Sjtcubergren-Larssons syndrom, 110 trichothyodystrophy,114 juvenil Gauchers sjukdom, 110 neutral lipidlagringssjukdom, Conradi-Htcubermann-Happle syndrom, Hays-Wells syndrom och ektodermal dysplasi115 kan också ibland förekomma som kollodionbarn. Membranet försvinner spontant hos 10% till 24% av nyfödda, för att ge plats för helt normal hud.110 116 tidigare beskrevs dessa fall som LI av den nyfödda, 117 men de kallas inte SHCB.118 vissa författare har föreslagit termen självförbättrande kollodioniktyos eftersom många av dessa patienter, när de granskas senare i barndomen eller som vuxna, har en varierande grad av anhidros och värmeintolerans och milda tecken på iktyos, såsom xeros och fin desquamation, särskilt i axillae och nacke.78
varken optisk mikroskopi eller ultrastrukturella undersökningar av kollodionbarn är specifika. Det är därför att föredra att fördröja hudbiopsin tills den definitiva fenotypen har utvecklats.
mutationer i generna TGM1,7,119aloxe3,78 och ALOX12B23,78,79 har identifierats hos patienter med SHCB. ALOX12B-mutationer är de vanligaste. I en serie av 15 skandinaviska patienter med SHCB hade 67% mutationer i ALOX12B-genen, 25% i ALOXE3-genen och 8,3% i tgm1-genen.78 mutationer hittades inte hos vissa patienter, och så kommer andra gener sannolikt också att vara inblandade. Det har spekulerats att dessa mutationer minskar enzymatisk aktivitet i livmodern men inte efter födseln.7 i livmodern, där det hydrostatiska trycket är högt, omvandlar kelation med vatten det muterade enzymet till en inaktiv konformation. Efter födseln, när trycket minskar, återgår enzymet till sin aktiva form och dess aktivitet ökar tillräckligt för att upprätthålla en normal eller minimalt påverkad fenotyp.7
Acral Self-healing Collodion Baby
även om collodion baby påverkar hela kroppen har fall begränsade till de akrala regionerna rapporterats. 1952, Finlay et al.120 rapporterade ett fall av kollodionmembran som endast påverkade händer och fötter och som följde en självläkande kurs. Nyligen har ett nytt fall av acral SHCB rapporterats i samband med mutationer av tgm1-genen.8 Det är inte känt varför dessa lesioner är begränsade till akrala regioner, även om faktorer associerade med platsberoende reglering av enzymaktivitet kan vara i drift.8
Baddräktiktyos
baddräktiktyos rapporterades först som en oberoende Arci-variant 2005 även om fall av iktyos med en speciell fördelning tidigare hade rapporterats.121-123 det har upptäckts främst hos patienter med sydafrikanskt ursprung, 9 även om det också har rapporterats hos individer från Europa och Medelhavsländerna.124 vid födseln har patienter ett generaliserat kollodionmembran som sedan skjul för att lämna den karakteristiska fördelningen av skalning. Stammen, den proximala regionen i armarna, inklusive axillan, nacken och hårbotten påverkas vanligtvis, medan den centrala delen av ansiktet, lemmarna och binjurregionen vanligtvis sparas.9 vågen är stora, lamellära och mörka i färg. Finare desquamation kan förekomma i popliteal och antecubital fossae.124,125 handflatorna och fotsulorna har mild diffus hyperkeratos medan ryggen på händer och fötter inte visar något engagemang.
histopatologisk studie av drabbad hud visar markerad hyperkeratos utan parakeratos, normala granulära skikt, mild eller måttlig akantos och ett milt lymfocytiskt infiltrat i övre dermis.9 observationer av elektronmikroskopi överensstämmer i de flesta fall med medfödd iktyos typ 2. Oinvolverad hud visar inga onormala fynd.124 125 i frisk hud reduceras Tgas 1-aktiviteten något och lokaliseras vanligtvis i pericellulära områden. I involverad hud är enzymatisk aktivitet kvarvarande och onormalt belägen i cytoplasman.124
mutationer har upptäckts i tgm1-genen hos alla patienter med baddräktiktyos som hittills studerats.119,124-126 den vanligaste mutationen är p.Arg315Leu, som har identifierats hos de flesta Sydafrikanska patienter och kan vara en grundande mutation. Oji et al.124 föreslog att hudtemperaturen kan spela en roll i utvecklingen av dessa manifestationer. Med hjälp av digital termografi visade författarna en stark korrelation mellan kroppstemperatur och desquamation, med de hetaste områdena i kroppen som de mest drabbade. Aufenvenne et al.127 visade en minskning av optimal temperatur för Tgas 1-aktivitet hos patienter med baddräktiktyos. Denna minskning observerades inte hos friska kontroller eller hos patienter med generaliserad LI. denna temperaturminskning skulle förklara fenotypen hos dessa patienter. Den optimala temperaturen är 37 CCB för det normala enzymet men 31 CCB för det muterade enzymet.
behandling
det primära syftet med behandlingen vid iktyos är att eliminera skalning och minska xeros utan att orsaka överdriven irritation (tabell 3). Innan beslut fattas om behandling bör aspekter som patientens ålder och kön, typ och svårighetsgrad av sjukdomen samt omfattning och plats för lesionerna beaktas.128
terapeutisk strategi vid autosomala recessiva medfödda Iktyoser.
| terapeutisk strategi för autosomala recessiva medfödda iktyoser | |
| badning och mekanisk eliminering av skalor | badning med natriumbikarbonat eller vetestärkelse, majsstärkelse eller risstärkelse; mekanisk avlägsnande av skalorna (1 eller 2 gånger om dagen) |
| topisk behandling (Sekventiell) | Ureainnehållande fuktgivarekeratinolytika med propylenglykolkombinerade keratinolytika (propylenglykol, sackaros-hydroxisyror eller urea)Keratinolytika i kombination med salicylsyraaktuella retinoiderhos nyfödda och små barn, applicera ett fordon utan aktiva ingredienser. Undvik urea, salicylsyra och mjölksyra på grund av risken för systemisk absorption |
| Oral behandling | orala retinoider (acitretin eller isotretinoin) |
| andra åtgärder | uppföljning av ektropion av oftalmologenregelbunden rengöring av ytterörat av öron-hals-nässpecialistenfysioterapi för att förhindra kontrakturer.Undvikande av ansträngande aktiviteter vid hög omgivningstemperaturhydroterapi |
badning och mekanisk eliminering av skalor
daglig badning rekommenderas för patienter med ARCI för att mekaniskt eliminera skalor och spår av fuktkräm. Detta är lättare om patienten är nedsänkt i vatten i 15 till 30minuter. Vissa författare rekommenderar att man tillsätter natriumbikarbonat i badet för att denaturalisera keratinerna och göra vattnet alkaliskt och så underlätta eliminering av skalorna.129 andra produkter som kan tillsättas inkluderar vetestärkelse, majsstärkelse eller risstärkelse. Badoljor är inte lämpliga eftersom de kan leda till ocklusion med efterföljande risk för bakteriell proliferation och försämring av termoregulering.
topisk behandling
fuktgivare och topiska keratolytiska medel är vanligtvis det första terapeutiska alternativet. De förbättrar hudbarriärfunktionen och underlättar desquamation. Milda lokala biverkningar, såsom övergående klåda, irritation eller stickande känsla kan uppstå.
natriumklorid, urea, vitamin E-acetat, glycerol och vaselin kan användas som fuktighetsgivare och smörjmedel. Hos patienter med tjock skalning och markerad hyperkeratos kan 1 eller flera keratolytiska medel, såsom XXL-hydroxisyror (mjölksyra och glykolsyra),130 salicylsyra, N-acetylcystein,131-133 urea (>5%),134 och propylenglykol tillsättas. Modulatorer av keratinocytdifferentiering används också. Dessa inkluderar aktuella retinoider (tretinoin, adapalen, tazaroten),135,136 kalcipotriol,137 och dexpanthenol.Aktuella retinoider orsakar ofta irritation och små, mycket smärtsamma sprickor.137 dessutom finns det en risk för absorption och teratogenicitet hos fertila kvinnor om de används för mycket.138 för att förbättra effektiviteten hos keratolytika och fuktighetsgivare kan ocklusiv förband appliceras i specifika områden som är eldfasta mot behandling.139 en additiv eller synergistisk effekt kan också uppnås genom att kombinera 2 eller flera keratolytiska medel eller fuktighetsgivare.140-142 behandling bör optimeras för varje individ, med tanke på tillståndets mycket varierande karaktär och hudkänslighet och skillnader som svar på varje behandling. Optimeringsprocessen kan hjälpas genom att behandla ena sidan av kroppen annorlunda än den andra för att möjliggöra jämförelser. Nyfödda och små barn ska behandlas med ett fordon utan några aktiva substanser eftersom huden är mycket fin och känslig och de flesta keratolytika tolereras inte. Dessutom är risken för perkutan absorption av topiska produkter såsom urea, salicylsyra och mjölksyra större.143-145
systemisk behandling
orala retinoider har keratolytiska effekter som hjälper till att eliminera skalor och förhindra överdriven hyperkeratos. Både isotretinoin och aromatiska retinoider (acitretin och etretinat) har visat sig vara effektiva vid behandling av ARCIs.128 146 147 Acitretin i en dos av 0,5 till 1 mg/kg/d är det mest använda läkemedlet, särskilt hos patienter med LI.148 patienter med CIE kan ha ett mer fullständigt svar och vid lägre doser.
de huvudsakliga biverkningarna är mukokutana störningar, teratogenicitet, muskuloskeletala störningar och onormal lipidprofil och transaminashöjning.149-152 när det gäller teratogenicitet, när det gäller etretinat och acitretin, bör läkemedlen undvikas under graviditet och patienter bör undvika att bli gravida i 3 år efter avslutad behandling.151 Isotretinoin har en kortare halveringstid och elimineras helt från organismen efter 1 månad och kan därför vara det föredragna alternativet hos kvinnor som vill bli gravida.128
behandlingsövervakningen bör omfatta ett laboratoriearbete med ett leverfunktionstest och lipidprofil innan behandlingen påbörjas, därefter vid 1 månad och var 3: e månad efter påbörjad behandling. Hos fertila kvinnor bör ett graviditetstest utföras under 2 veckor innan behandlingen påbörjas och en effektiv preventivåtgärd bör användas från 4 veckor före behandling till 3 år därefter (i fallet med acitretin). När långvarig behandling krävs med retinoider, bör tillväxt och benutveckling övervakas. Vissa författare föreslår att man utför en benstudie före behandling följt av en årlig undersökning.151 nya riktlinjer rekommenderar inte att du utför rutinmässig radiografi på grund av eventuella skadliga effekter.152 istället rekommenderas selektiva radiografiska studier hos patienter som har atypisk bensmärta.152
ett alternativ till systemisk retinoidbehandling är användningen av läkemedel som kallas retinsyrametabolismblockerande medel, vilket ökar de endogena nivåerna av retinsyra. Ett sådant läkemedel är liarozol, som har fått föräldralös status för behandling av LI, CIE och HI av Europeiska läkemedelsmyndigheten och US Food and Drug Administration.153-155 detta läkemedel har visat sig vara effektivare än acitretin i kliniska prövningar och det tolereras också bättre och har en bättre farmakokinetisk profil.154
annan medicinsk vård
hos patienter med ektropion kan applicering av artificiella tårar och ögonsmörjmedel och fuktgivande ansiktet i ansiktet och kinderna i synnerhet minska palpebral retraktion. Kirurgisk korrigering är ett giltigt alternativ i svåra fall, men detta måste vanligtvis upprepas några år senare. Hydroterapi kan vara fördelaktigt.156 patienter bör rådas att undvika ansträngande fysisk aktivitet när omgivningstemperaturen är hög, eftersom hypohidros medför risken för värmeslag och kramper. Orala retinoider kan förbättra termoreguleringen.157 fysioterapi är viktigt för att förhindra flexionskontrakt, särskilt när det gäller HI. Regelbunden rengöring av den yttre hörselkanalen av en öron-hals – nässpecialist kan förhindra att skalor ackumuleras och därmed förhindra hörselnedsättning.
genetisk rådgivning och Prenatal diagnos
när en patient diagnostiseras med iktyos bör han eller hon erbjudas lämplig genetisk rådgivning där arten av störningen, överföringsläget och risken för framtida manifestationer i familjen förklaras. Prenatal diagnos kan indikera om fostret påverkas och om så är fallet kan psykologisk förberedelse av familjen erbjudas och problem förväntas under graviditet och födelse. Föräldrarna kan ges möjlighet till abort Om ingen behandling är tillgänglig. Dessutom, om genterapi för dessa tillstånd blir tillgänglig i framtiden, skulle prenatal diagnos möjliggöra tillämpning av denna terapi så tidigt som möjligt.
i mer än 20 år utfördes prenatal diagnos genom att ta ett biopsiprov av fosterhud och studera det genom optisk mikroskopi, elektronmikroskopi eller immunhistokemi.158 159 denna invasiva procedur kunde endast utföras i de sena faserna av graviditeten, mellan veckorna 15 och 23 av graviditeten, och var förknippad med en 1% till 3% risk att förlora fostret.160,161 identifieringen av de molekylära mekanismerna för ärftliga hudsjukdomar har möjliggjort en mycket tidigare diagnos baserad på genetiska tekniker.102,162–164 fetalt DNA erhålls genom fostervattensprov som utförs mellan veckorna 15 och 20 eller genom kororisk villusprovtagning mellan veckorna 10 och 12. Risken för fosterförlust med dessa tekniker är mindre än mellan 0,5% och 1%.165 andra icke-invasiva metoder i utveckling är analys av fostercell-DNA och fritt foster-DNA i moderns cirkulation166 samt användning av 3-dimensionell ultraljud.167,168
preimplantationsgenetisk diagnos kan också vara möjlig i in vitro fertiliseringstekniker, så att endast befruktade ägg som är fria från mutationen implanteras i livmodern och därigenom undviker behovet av abort i de flesta fall.169
framtida strategier för genetisk behandling av iktyos
även om viktiga framsteg har gjorts i den genetiska diagnosen av iktyos, bedrivs också nya strategier för dessa sjukdomar.170 huden är det mest tillgängliga organet för genöverföringsterapier, och så är sådana tekniker minimalt invasiva.171 huden har emellertid också unika immunologiska egenskaper som inte gynnar långsiktigt uttryck av en transgen produkt.172 i LI lyckades en process med ex vivo-genöverföring återställa normalt tgm1-uttryck och korrigera fenotypen av hud transplanterad på baksidan av immunsupprimerade möss.173,174 nyligen har fenotypen av odlade keratinocyter från patienter med HI på grund av mutationer i ABCA12-genen också återhämtats.3
intressekonflikter
författarna förklarar att de inte har några intressekonflikter.