Frontiers in Neurology
introduktion
Neuroacanthocytosis är en överordnad term för en grupp sällsynta syndrom som kännetecknas av neurologiska symtom i kombination med spikiga deformerade röda blodkroppar (acanthocytes). Denna rapport fokuserar på Chorea-acanthocytosis (ChAc), som är en orphan sjukdom med uppskattade 1,000-drabbade individer över hela världen orsakade av autosomal-recessiva mutationer i vps13a (vacuolar protein sorting 13 homolog a) gen på kromosom 9q (1). Sjukdomen löper en progressiv kurs, orsaker till ökad dödlighet kan minska motoriska funktioner som korrelerar med riskförhållanden som dysfagi, men också plötsliga oväntade dödsfall har beskrivits (2, 3). Hittills finns det ingen orsakande behandling.
den misstänkta samexistensen av akantocytos och rörelsestörningar rapporterades först på 1970-talet av Levine och Critchley (4, 5) och har sedan dess accepterats som den framträdande egenskapen hos sjukdomen. Men med vår fallrapport beskriver vi en sällsynt klinisk fenotyp av ChAc som saknar uppenbara rörelsestörningar, vilket tyder på ett bredare utbud av kliniska presentationer. Diagnostiska verktyg måste möta dessa utmaningar för att fastställa rätt diagnos föreslår vi här molekylär neuroimaging som en nyckelmetod.
fallrapport
den manliga indexpatienten presenterades först vid 25 års ålder med två oprovocerade bilaterala toniska kloniska anfall. Det fanns ingen medicinsk historia. Hjärnan MR-skanning och EEG vid den åldern var normala och ingen behandling initierades på grund av patientens motvilja och sällsynta händelser. Patienten hade dock pågående anfall som nu presenterades som partiell epilepsi. Han beskrev återkommande känslor av plötslig yrsel som diagnostiserades som en svindlande aura. Dessutom hade han dyskognitiva anfall där han antingen visade (a) svarlöshet och recitering av turkiska böner, (b) muntliga automatismer, eller (c) fiddling med händerna och yttrade ljud—allt delvis utvecklas till toniska kloniska anfall. Video-EEG visade nu lokala spikvågskomplex både i höger och vänster temporal lob. I enlighet med anfallets semiologi ställdes diagnosen bilateral temporal lobepilepsi och medicinering med levetiracetam startades.
vid en ålder av 31 anfall undertrycktes inte helt, som en del av ytterligare diagnostik av temporal lobepilepsi genomgick patienten en FDG-PET (Figur 1) som visade en bilateral mesiotemporal hypometabolism, i linje med mesiotemporal anfall ursprung. Det fanns emellertid också en markant, mycket ovanlig striatal hypometabolism som väckte misstanken om en neurodegenerativ rörelsestörning. Följaktligen inleddes en omfattande uppföljningsutvärdering (se Figur 2). En FP-CIT-SPECT utfördes som gav en måttligt bilateral förlust av tillgänglighet för striatal dopamintransportör, i linje med en minskning av nigrostriatal integritet (Figur 1). En högupplöst Mr presenterade nu bilateral atrofi av nucleus caudatus (Figur 3). Neurologisk undersökning visade ett diskret bundet gångmönster och hypomimi men ingen annan tillgivenhet av det extrapyramidala motorsystemet och inga bulbarsymtom som matningsdystoni. Bortsett från låg reflexstatus på nedre extremiteterna, för vilka nervledningsstudier bekräftade en sensorimotorisk axonal polyneuropati, var den neurologiska undersökningen normal. Neuropsykologiska symtom inkluderade aspekter av en ångestsyndrom och patienten rapporterade svårigheter med korttidsminnesförlust. Neuropsykologisk testning bekräftade emellertid inte uppenbart mnestic-fel utan snarare en ospecifik distraktion, som dessutom kan ha förbättrats av otillräcklig anfallssuppression vid den tiden. Laboratorietester var negativa för symtomatisk epilepsi (dvs., autoimmun-encefalitantikroppar, antineuronala antikroppar). Cerebrospinalvätska visade inga tecken på inflammation men en måttlig proteinökning (765 mg/l). Analys av biogena aminer i cerebrospinalvätskan avslöjade en höjning av glutamin som vi associerade med nya anfall. I perifert blod hittades 0,4% akantocyter. Serologisk testning för Wilsons sjukdom var negativ. Märkbar var en ihållande höjning av kreatinkinas (intervall: 1 000-3 000 U/l) vilket ledde till misstänkt diagnos av en mitokondriell störning. För ytterligare undersökningar utfördes ett ischemiskt laktattest som visade normala resultat. En muskelbiopsi av M. gastrocnemicus utfördes, men analys av mitokondriell funktion och andningskedja var normal.
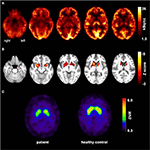
Figur 1. Resultat av molekylära avbildningsstudier. Transaxial FDG-PET-bilder (a) visade inte bara en mild bilateral mesiotemporal hypometabolism (i linje med mesiotemporal anfall ursprung) men också en markant striatal hypometabolism som visade sig vara mycket signifikant jämfört med friska kontroller . En ytterligare FP-CIT-SPECT-undersökning (C) avslöjade också en måttlig förlust av striatala dopamintransportörer, vilket bevittnade en minskning av nigrostriatal integritet (en åldersmatchad hälsosam kontroll visas för jämförelse; DVR, distributionsvolymförhållande).

Figur 2. Klinisk och diagnostisk kurs hos indexpatienten.
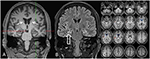
Figur 3. MR visar diskret bilateral caudathuvudatrofi och en mindre och hyperintensiv högersidig hippocampus som indikerar högersidig hippocampus skleros . Den vänstra hippocampus är något hyperintensiv men inte atrofisk. Bilateral caudathuvudatrofi bekräftas med CVR-analys, där blå färger visar minskad grå och röd färger ökad cerebrospinalvätskevolym respektive (C).
familjehistoria avslöjade en konsanguinitet hos sina föräldrar (första kusiner), som själva inte hade någon signifikant medicinsk historia. Av hans sex syskon hade två (mellan 20-30 år) nu också drabbats av allmänna anfall (se Figur 4). Medicinska register över hans syskon var inte tillgängliga för oss.
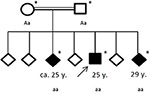
Figur 4. Stamtavla av den drabbade familjen, med ålder av första epileptiska anfall i år (y). Pil: index patent. Asterisk: genetisk testning utförd. Aa, heterozygot; aa, homozygot för c. 4326 T> A (S.Tyr1442*).
under hypotesen om en ärftlig neurodegenerativ rörelsestörning hänvisades patienten och hans familj för genetisk testning. Exome-sekvensering avslöjade en homozygot roman stympande mutation c.4326 T>A (S.Tyr1442*) i ChAc-genen VPS13A hos de tre drabbade syskonen. De konsanguine föräldrarna upptäcktes båda heterozygota. Inga andra varianter eller mutationer identifierades i exome-sekvenseringen.
faktum är att i en uppföljning vid 33 års ålder hade indexpatienten fortfarande bara marginella extrapyramidala symtom och ingen var närvarande i någon av hans systrar. Han gav nu intrycket av lätt styvhet endast under priming på höger övre extremitet och en mild bradykinesi i övre extremiteterna. Under behandling med levetiracetam och lakosamid har patienten varit anfallsfri.
diskussion
med det aktuella fallet visar vi att ChAc kan kliniskt manifestera med temporal lobepilepsi som saknar några signifikanta motoriska symtom på grund av en ny motsvarande mutation i vps13a-genen.
sjukdomsförloppet kännetecknas vanligtvis av en progressiv rörelsestörning (inklusive chorea, dystoni, parkinsonism), kognitiva och psykiatriska förändringar och myopatiska symtom med serologiska markörer för akantocytos och hyperckemi. Kramper har tidigare noterats som ett symptom på ChAc (6) men rörelsestörningar anses fortfarande vara det viktigaste symptomet. Bevis uppstår att epilepsi, och mer specifikt epilepsi med ursprung i den temporala loben, kan vara en underskattad fenotyp av ChAc. Peluso et al. rapporterade en patient som liknar vår, som, även vid 46 års ålder, inte visade rörelsestörningar medan de presenterade neuroimaging-fynd som indikerar basal ganglia-engagemang (7). I enlighet med det, Scheid et al. beskrev tre patienter som led av mesial temporal lobepilepsi och skleros (baserat på MR-studier) som det dominerande symptomet och diagnostiserades med ChAc (8). Dessutom, Mente et al. nyligen gav obduktionsresultat av en ChAc-patient som visade inte bara basal ganglia atrofi utan också hippocampal skleros (9). I linje med vårt fall verkar bilateralt engagemang av hippocampus vara ett vanligt resultat (10, 11). Det exakta förhållandet mellan anfall och skleros är dock fortfarande en fråga om höna eller ägg. Den korrelerande rollen av chorein i den strukturella utvecklingen av hippocampus är ännu inte helt förstådd. Intressant, i en musmodell av ChAc strukturell förändring sågs, som hippocampal proteinuttryck av GABA (A) receptorn gamma 2 och dess förankringsprotein chorein ökades (12).
i klinisk rutin kan denna fenotyp fortfarande vara utmanande för att hitta rätt diagnos. Den första avgörande ledtråden mot en neurodegenerativ sjukdom hos vår patient var en framträdande minskning av striatal glukosmetabolism och en måttlig minskning av nigrostriatal integritet på FDG-PET respektive FP-CIT-SPECT. Den lilla samlingen av funktionella avbildningsresultat, baserat på fallseriebeskrivning, visar att förändringar i metabolism och dopaminerg dysfunktion oftast förekommer i caudatkärnan och putamen (13). Hjärnan MR avslöjade atrofi av kärnan caudatus under sjukdomsförloppet, vilket har beskrivits som ett typiskt fynd i ChAc (14). Följaktligen inkluderar distinkta neurologiska egenskaper vanligtvis chorea, matningsdystoni, orofaciolingual dyskinesier och tics, tillsammans med andra symtom på basal ganglia-tillgivenhet såsom parkinsonism och dystoni (3). Det är frestande att spekulera i att de observerade striatala förändringarna hos vår patient fortfarande var under tröskeln för att orsaka symtom (dvs prodromala avbildningsfynd), vilket i slutändan kan uppstå när sjukdomen fortskrider. Vi uppmuntrar därför strukturell och molekylär avbildning som ett känsligt diagnostiskt verktyg i denna föräldralösa sjukdom.
i ChAc finns beskrivningar av akantocytantal mellan 5 och 50% (3), men det finns några fallrapporter som visar att akantocyter kan förekomma först sent under sjukdomsförloppet (15), kan vara mycket låga eller till och med vara helt frånvarande (16). Därför bör akantocyter inte anses vara obligatoriska för att göra diagnosen. Dessutom kan epilepsi fördröja diagnosen eftersom den döljer andra typiska symtom på ChAc. I synnerhet kan psykiatriska symtom som personlighetsförändringar och kognitiv försämring, som är mycket vanliga, misstolkas och tillskrivas läkemedelsrelaterade biverkningar (dvs. av levetiracetam) eller kramprelaterade. Sekundär hyperckemi ses efter allmänna toniska kloniska anfall och en ihållande höjning kan förbises.
vårt fall belyser också den avgörande fördelen med exome-sekvensering för att upprätta en korrekt diagnos, särskilt vid sällsynta sjukdomar eller när symtomen är vaga. Familjen av däggdjur vps13 (A–D) gener är av stigande intresse och mutationer har identifierats i andra neurologiska sjukdomar såsom autosomal recessiv Parkinsons sjukdom eller autosomal recessiv Spinocerebellär ataxi (17). Recessiva mutationer i VPS13D orsakar rörelsestörningar i barndomen (18). Den underliggande patofysiologin av ChAc är ännu inte fullständigt dechiffrerad, men det visades att VPS13A kodar proteinet chorein som uttrycks allestädes närvarande i hjärnan och i ett stort antal andra vävnader (19). Studier visar att det deltar i signalvägar som reglerar cytoskeletal arkitektur, exocytos och cellöverlevnad (20). En annan mutation som har beskrivits för att presentera en anfallsdominerad fenotyp hos 9 patienter med ChAc är C.2343del-mutationen i vps13a-genen (21). Det är rimligt att anta att specifika mutationer i samma gen resulterar i en viss avvikelse av chorein som orsakar en unik fenotyp, till exempel genom att påverka olika delar av hjärnan. Den underliggande patofysiologin är dock fortfarande bara spekulativ och ytterligare dokumentation av de orsakande mutationerna kommer att vara av intresse. Vi visar här att den trunkerande mutationen c.4326 T>A (S.Tyr1442*), som inte har beskrivits tidigare, resulterar i en liknande fenotyp med dominerande epilepsi hos alla drabbade familjemedlemmar.
avslutande anmärkningar
epilepsi (särskilt bilateral temporal lobepilepsi) är sannolikt en dominerande fenotyp av ChAc. Därför bör en sökning efter ytterligare röda flaggor (som hyperckemi, familjehistoria, polyneuropati) utföras noggrant. I misstänkta fall söka efter akantocyter i blodet och hjärnan MRI bör läggas eftersom dessa verktyg är allmänt tillgängliga. Om du är osäker bör diagnostik kompletteras med FDG-PET och/eller FP-CIT-SPECT eftersom de är känsliga för att upptäcka striatal involvering. Vid autosomala recessiva epilepsier med indikativa fynd för en neurodegenerativ sjukdom bör VPS13A singelgentest eller chorein Western blot sedan användas för att fastställa rätt diagnos. Det senare bör också övervägas i andra neurodegenerativa störningar utan genetiskt definierad etiologi.
Etikuttalande
skriftligt informerat samtycke erhölls från patienten för deltagande i studien. Skriftligt informerat samtycke erhölls från patienten för publicering av denna fallrapport.
Författarbidrag
JW, MR och SK deltog i patienthantering. JW skrev det första utkastet till manuskriptet. LF, HU, och PM beredda siffror. Alla författare bidrog till manuskriptrevision, läste och godkände den inlämnade versionen.
intressekonflikt uttalande
HU är aktieägare i Veobrain Gmbh, en spin-off av University Medical Center Freiburg.
de återstående författarna förklarar att forskningen genomfördes i avsaknad av kommersiella eller finansiella relationer som kan tolkas som en potentiell intressekonflikt.
1. Han är en av de mest populära och mest populära. Neuropsykiatri av neuroacanthocytosis syndrom. Neurosci Biobehav Rev. (2011) 35:1275-83. doi: 10.1016/j.neubiorev.2011.01.001
PubMed Abstrakt / CrossRef Fulltext / Google Scholar
2. Walker RH. Hantering av neuroacanthocytosis syndrom. Tremor Andra Hyperkinet. Mov. (2015) 5:346. doi: 10.7916 / D8W66K48
PubMed Abstrakt / CrossRef fulltext / Google Scholar
3. Velayos Baeza A, Dobson-sten C, Rampoldi L, Bader B, Walker RH, Danek A, et al. Chorea-Akantocytos. GeneReviews. Seattle, WA: University of Washington (1993).
4. Critchley EM, Clark DB, Wikler A. Akantocytos och neurologisk störning utan Betalipoproteinemi. Arch Neurol. (1968) 18:134–40.
PubMed Abstrakt
5. Levine IM, Estes JW, Looney JM. Ärftlig neurologisk sjukdom med akantocytos. Ett nytt syndrom. Arch Neurol. (1968) 19:403–9.
PubMed Abstrakt / Google Scholar
6. Hardie RJ, Pullon HW, Harding AE, Owen JS, Pires M, Daniels GL, et al. Neuroakanthocytos. en klinisk, hematologisk och patologisk studie av 19 fall. Hjärna(1991) 114 (Pt 1A): 13-49.
PubMed Abstrakt / Google Scholar
7. Vi har ett stort utbud av produkter och tjänster. Chorea-akantocytos utan chorea: utvidgning av den kliniska fenotypen. Parkinson Relat Disord. (2017) 41:124–6. doi: 10.2016/J.parkreldis.2017.05.013
PubMed Abstrakt / CrossRef Fulltext / Google Scholar
8. Scheid R, Bader B, Ott DV, Merkenschlager a, Danek A. utveckling av mesial temporal lobepilepsi i chorea-akantocytos. Neurologi (2009) 73:1419-22. doi: 10.1212 / WNL.0b013e3181bd80d4
PubMed Abstrakt / CrossRef fulltext / Google Scholar
9. Mente K, Kim SA, Grunseich C, Hefti MM, Crary JF, Danek A, et al. Hippocampal skleros och mesial temporal lobepilepsi i chorea-acanthocytosis: ett fall med klinisk, patologisk och genetisk utvärdering. Neuropathol Appl Neurobiol. (2017) 43:542–6. doi: 10.1111/nan.12403
PubMed Abstrakt / CrossRef Fulltext / Google Scholar
10. Bader B, Vollmar C, Ackl N, Ebert a, la Foug C, Noachtar S, et al. Bilateral temporal lobepilepsi bekräftad med intrakraniell EEG vid chorea-akantocytos. Beslag (2011) 20:340-2. doi: 10.1016/j.beslag.2010.12.007
PubMed Abstrakt / CrossRef Fulltext / Google Scholar
11. Marson AM, Bucciantini E, Gentile E, Geda C. Neuroacanthocytosis: kliniska, radiologiska och neurofysiologiska fynd i en italiensk familj. Neurol Sci. (2003) 24:188–9. doi: 10.1007 / s10072-003-0123-1
PubMed Abstrakt / CrossRef fulltext / Google Scholar
12. Det är en av de mest populära och mest populära. Choreinbrist leder till uppreglering av gephyrin och GABA(A) receptor. Biochem Biophys Res Commun. (2006) 351:438–42. doi: 10.1016 / j. bbrc.2006.10.070
PubMed Abstrakt / CrossRef Fulltext / Google Scholar
13. Ehrlich DJ, Walker RH. Funktionell neuroimaging och chorea: en systematisk granskning. J Clin Mov Disord. (2017) 4:8. doi: 10.1186 / s40734-017-0056-0
PubMed Abstrakt / CrossRef fulltext / Google Scholar
14. Connolly BS, Hazrati L – N, Lang AE. Neuropatologiska fynd i chorea-acanthocytosis: nya insikter i mekanismer som ligger bakom Parkinsonism och anfall. Acta Neuropathol. (2014) 127:613–5. doi: 10.1007 / s00401-013-1241-3
PubMed Abstrakt / CrossRef fulltext / Google Scholar
15. Sorrentino G, de Renzo A, Miniello S, Nori O, Bonavita V. sen utseende av akantocyter under chorea-akantocytos. J Neurol Sci. (1999) 163:175–8.
PubMed Abstrakt / Google Scholar
16. Bayreuther C, Borg M, Ferrero-Vacher C, Chaussenot A, Lebrun C. Chor-acanthocytose sans acanthocytes. Revue Neurol. (2010) 166:100–3. doi: 10.1016 / j.neurol.2009.03.005
CrossRef Fulltext / Google Scholar
17. Lesage S, Drouet V, Majounie E, Deramecourt V, Jacoupy M, Nicolas A, et al. Förlust av VPS13C-funktion vid autosomal-recessiv Parkinsonism orsakar mitokondriell dysfunktion och ökar PINK1/Parkin-beroende mitofagi. Am J Hum Genet. (2016) 98:500–13. doi: 10.1016/j.ajhg.2016.01.014
PubMed Abstrakt / CrossRef Fulltext / Google Scholar
18. Han är en av de mest kända i världen. Recessiva mutationer i > VPS13D orsakar rörelsestörningar i barndomen. Ann Neurol. (2018) 83: I6. doi: 10.1002 / ana.25204
PubMed Abstrakt / CrossRef Fulltext / Google Scholar
19. Det är en av de mest populära och mest populära. In vivo distribution och lokalisering av chorein. Biochem Biophys Res Commun. (2007) 353:431–5. doi: 10.1016 / j. bbrc.2006.12.059
PubMed Abstrakt / CrossRef Fulltext / Google Scholar
20. Lang F, Pelzl L, Schobyls L, Hermann A, Fbyler M, Schobylffer TE, et al. Neuroner, erytrocyter och bortom-choreins olika funktioner. Neurosignaler (2017) 25:117-26. doi: 10.1159/000485457
PubMed Abstrakt / CrossRef fulltext / Google Scholar
21. Han är en av de mest kända och mest kända i världen. Kramper som presenterar och framträdande symptom i chorea-acanthocytosis med C. 2343del VPS13A genmutation. Epilepsi (2016) 57:549-56. doi: 10.1111 / epi.13318
PubMed Abstract | CrossRef Full Text | Google Scholar