Genetiken för klyftläpp och gommen
 Intro/abstractCleft lip med eller utan klyftgom är en komplex medfödd anomali som kan isoleras eller ses tillsammans med andra missbildningar. Det kan också vara en del av fenotypen av ett genetiskt syndrom. Denna artikel fungerar som en översyn av förekomsten av läpp-och gomspalt, risker för återfall och risker för andra medfödda anomalier. Genetiska syndrom och teratogena exponeringar som är kända för att vara associerade med orala klyftor kommer att undersökas. Dessutom genetiska tester som vanligtvis begärs i pediatrisk klinisk genetik inställning för utvärdering av patienten med klyvläpp och gom kommer att diskuteras.
Intro/abstractCleft lip med eller utan klyftgom är en komplex medfödd anomali som kan isoleras eller ses tillsammans med andra missbildningar. Det kan också vara en del av fenotypen av ett genetiskt syndrom. Denna artikel fungerar som en översyn av förekomsten av läpp-och gomspalt, risker för återfall och risker för andra medfödda anomalier. Genetiska syndrom och teratogena exponeringar som är kända för att vara associerade med orala klyftor kommer att undersökas. Dessutom genetiska tester som vanligtvis begärs i pediatrisk klinisk genetik inställning för utvärdering av patienten med klyvläpp och gom kommer att diskuteras.
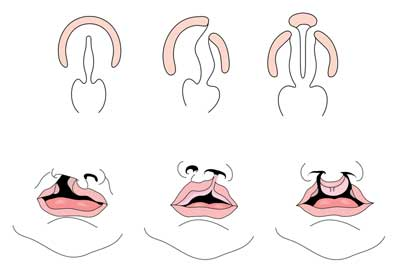
kluven läpp med eller utan gomspalt (CL/CP) skiljer sig från en isolerad gomspalt (CP) på embryonala, epidemiologiska och genetiska nivåer. Kluven läpp resulterar vanligtvis från maxillär framträdande och medial nasal framträdande misslyckas med att smälta mellan den femte och sjätte veckan av embryonal utveckling. Normal gomutveckling är resultatet av bildandet av den primära gommen och den sekundära gommen. Den primära gommen bildas vid veckor sex till sju genom utveckling och fusion av mediala nasala, laterala nasala och maxillära processer. Den sekundära gommen härstammar från palatalhyllorna (som utvecklas från de parade maxillära processerna i den första grenbågen) och blir horisontella och smältande och bildar de hårda och mjuka gommen runt den nionde veckan av embryonal utveckling. Hyllor smälter också med den primära gommen och nässeptumet. (1)
orala klyftor är en av de vanligaste fosterskadorna som ses i neonatal plantskola, med en total prevalens av 1.6 per tusen nyfödda över hela världen, med CL/CP sett i ungefär en per tusen födda och CP sett i 0.6 per tusen födda. (2) Det finns en högre frekvens av CL/CP hos individer av Asiatisk, afrikansk och indiansk härkomst. CL / CP är också vanligare hos män. Däremot finns det ingen signifikant skillnad i förekomst av CP bland olika etniska bakgrunder, och CP är vanligare hos kvinnor. (3) risker för återfall inom en familj beror på om klyftan är isolerad (utan några andra kliniska fynd) eller ses som en del av ett genetiskt syndrom. De flesta fall av orala klyftor isoleras (cirka 80%). Isolerade klyftor tros ha multifaktoriell arv: de beror på en kombination av flera faktorer, både genetiska och miljömässiga. Risken för återfall (Tabell 1) ökar när det finns mer än en drabbad släkting. Risken för återfall ökar också ju allvarligare defekten är.

Klyvläpp och gom kan ses med andra medfödda anomalier. Sannolikheten för en genetisk eller teratogen etiologi ökar de mer medfödda anomalierna som en patient presenterar. Förekomst av andra problem som intellektuell funktionsnedsättning, beteendeproblem som autism, dysmorfiska Egenskaper eller andra medicinska problem kommer också att göra en genetisk störning eller en teratogen exponering mer sannolikt. Cirka 13% av individer med kluven läpp kommer att ha andra medicinska problem eller avvikelser. Antalet ökar till 37% Med klyftläpp och gom och till 47% Med klyftgom ensam.
Prenatal exponering för teratogena medel (såsom talidomid, antikonvulsiva medel, alkohol, retinsyra och cigaretter) och modersjukdom (såsom diabetes, rubella och folatbrist) har visat sig öka risken för orala klyftor. Närvaron av amniotiska band ökar också risken för klyftor. Perikonceptuell folsyratillskott är känt för att minska risken för orala klyftor.
Pierre Robin sequence är en kraniofacial anomali som kännetecknas av mandibulär hypoplasi eller mikrognathia, sekundär U-formad klyftgom och glossoptos som leder till obstruktiv apnea och matningssvårigheter. Pierre Robin sekvens kan ses som en del av genetiska syndrom (22q11.2 deletionssyndrom, Stickler syndrom; beskrivs nedan). (5)
det finns hundratals genetiska syndrom associerade med orala klyftor, inklusive cytogenetiska avvikelser (aneuploidier, mikrodeletioner) och störningar med en gen (Mendelian). Att bekräfta en genetisk diagnos är avgörande för att bestämma prognosen och skapa en risk för återfall.
Aneuploidier som Trisomi 13 och 18 har en stark koppling till CL/CP. Trisomi 13 (AKA Patau syndrom) är associerat med tre kopior av kromosom 13 eller obalanserade Robertsonian-translokationer som involverar kromosom 13. Spädbarn födda med detta tillstånd dör vanligtvis under nyföddperioden. Kliniska funktioner inkluderar kluven läpp och gom, tillväxthämning, allvarliga centrala nervösa missbildningar (inklusive holoprosencephaly), mikrocefali, mikroptalmi, iris coloboma, frånvaro av ögonen, missbildade öron, polydactyly, knutna nävar, vippbotten Fötter, medfödda hjärtfel och urogenitala defekter. Mittlinjeklyftor (annars mycket sällsynta) kan ses vid trisomi 13 på grund av risken för mittlinjedefekter, inklusive holoprosencephaly. Trisomi 18 (AKA Edwards syndrom) beror vanligtvis på tre distinkta kopior av kromosom 18 och är associerad med dåligt postnatalt resultat. Kliniska funktioner inkluderar kluven läpp och gom, utvecklingsstörning, underlåtenhet att frodas, medfödd hjärtsjukdom, hypertoni, micrognathia, kort bröstben, låg uppsättning missbildade öron, knutna händer, rocker botten fötter, och hypoplastiska naglar, bland andra. Trisomi 13 och 18 kan enkelt bekräftas eller uteslutas genom att göra kromosomanalys (karyotypning).
Mikrodeletionssyndrom involverar vanligtvis radering av en del av en kromosom. Dessa borttagningar kan vara för små för att detekteras genom standardkaryotypning och kan kräva att FISH (fluorescens in situ hybridisering) eller microarray-teknik detekteras. Ett välkänt mikrodeletionssyndrom associerat med gomspalt är 22q11.2 deletionssyndrom (aka Digeorge/Velocardiofacial syndrom). Palatala abnormiteter inklusive velofaryngeal inkompetens, submukosala klyftor, bifid uvula och klyftgom ses hos 69% av individerna med 22q11.2 deletion och kan vara en del av Pierre Robin-sekvensen. Andra kliniska fynd inkluderar medfödd hjärtsjukdom, hörselnedsättning, dysmorfa egenskaper, immunbrist, hypokalcemi, njuranomalier, utfodringsproblem, skelettanomalier och psykiatriska störningar. Cirka 10% av fallen av 22q11.2 deletionssyndrom tros vara familjärt. Borttagningen segregerar på ett autosomalt dominerande sätt.(6) Wolf-Hirschhorn-syndrom, som beror på en deletion i den korta armen av kromosom 4, är också associerad med orala klyftor (hos 25% till 50% av de drabbade individerna). Karakteristiska ansiktsdrag (inklusive framträdande glabella som leder till” grekisk krigare hjälmutseende”), medfödd hjärtsjukdom, intellektuell funktionsnedsättning, kramper, misslyckande att trivas, mikrognathia, preaurikulära taggar eller gropar och hypodonti kan också ses som en del av tillståndet.(7)
enstaka genstörningar med orala klyftor inkluderar Stickler syndrom, Treacher Collins syndrom och Van der Woude syndrom, bland många andra. Stickler syndrom är en kollagenstörning med autosomalt dominerande och, mindre vanligt, autosomalt recessivt arv. Vanliga funktioner inkluderar gomspalt (ses som en del av Pierre Robin sekvens eller utan micrognathia), hörselnedsättning (sensorineural och ledande), skelett fynd (tidig debut artrit, spondyloepiphyseal dysplasi), okulära anomalier (hög närsynthet, glaskroppen avvikelser) och karakteristiska ansiktsdrag (med underutveckling av överkäken och nasal bro, midface retrusion). Genetisk testning för Stickler syndrom kan vara komplex, eftersom mutationer i minst sex gener har beskrivits hos drabbade individer. Cirka 90% av patienterna med Stickler syndrom har mutationer i COL2A1-genen och har en autosomal dominerande form av tillståndet.(8) Treacher Collins syndrom är ett autosomalt dominerande tillstånd som kännetecknas av gomspalt med eller utan läpp i 28% av de drabbade individerna. Andra avvikelser inkluderar hypoplasi hos de zygomatiska benen och underkäken, yttre öronanomalier, kolobom i det nedre ögonlocket, ledande hörselnedsättning, frånvaro av nedre ögonfransar, preaurikulär hårförskjutning på kinderna och choanal stenos eller atresi. Diagnosen av Treacher Collins syndrom är baserad på kliniska och radiografiska fynd. Mutationer i minst tre gener har beskrivits, med mutationer i TCOF1 sett hos 78% till 93% av patienterna.(9) Van der Woude syndrom kännetecknas av närvaron av medfödda, vanligtvis bilaterala, paramedianska fistlar i underläppen (gropar), eller ibland små högar med en sinuskanal som leder från en slemhinnor i läppen och orala klyftor (inklusive CL/CP och CP). Van der Woude är ett autosomalt dominerande tillstånd associerat med mutationer i IRF6-genen (10). Testning för single-gen eller multi-gen villkor kräver direkt analys av genen genom sekvensering och/eller deletion/duplicering analys (såsom MLPA).
med tanke på att genetiska syndrom med klyvläpp och gom kan associeras med aneuploidier, kromosommikrodeletioner/mikroduplikationer eller enstaka genstörningar kan genetisk testning vara en komplicerad process. En grundlig sjukdomshistoria, en tre generationens stamtavla, en graviditetshistoria och en dysmorfologiundersökning av en klinisk genetiker kan klargöra den kliniska bilden och möjliggöra riktad genetisk testning. Nyare tekniker inklusive microarray möjliggör identifiering av små mikrodeletioner och mikroduplikationer som tidigare missats av standardkaryotypning. Tyvärr leder denna teknik också till identifiering av raderingar och duplikationer av okänd klinisk betydelse, vilket komplicerar den genetiska rådgivningsprocessen. Testning för engenstörningar eller Mendeliska störningar kräver klinisk tillgänglighet av genetisk testning för den önskade genen. Det kan också vara dyrt om det inte täcks av sjukförsäkring. Ny teknik som nästa generations sekvensering, exome-sekvensering eller genomsekvensering (känd kollektivt som genomiska tester) har nu blivit kliniskt tillgängliga. Genom att analysera hundratusentals gener samtidigt ökar dessa tester signifikant diagnostisk kraft och utbyte. Jämfört med andra tekniker kan dessa tester ge ett svar snabbare och på ett mer kostnadseffektivt sätt. Inom forskningsområdet har exome och genomsekvensering lett till identifiering av nya gener samt expansion av kliniska egenskaper och spektrum för genetiska mutationer. Som med microarray-teknik kan genomiska tester upptäcka syndrom som inte är relaterade till patientens presentation och/eller anledning till testning. Med tanke på den inneboende komplexiteten i genetisk testning, informerat samtycke är nödvändigt.
slutsats
även om klyvläpp och gom är en isolerad anomali i de flesta fall finns det en stark koppling mellan orala klyftor och andra anomalier och genetiska syndrom. En genetisk utvärdering av en klinisk genetiker och en genetisk rådgivare är avgörande för förväntad vägledning och för att bestämma risker för återfall. Genetisk testning, som kräver informerat samtycke, kan samordnas och tolkas under en genetisk utvärdering.
Anya Revah, MS, är senior genetisk rådgivare vid Avdelningen för medicinsk genetik vid Maimonides spädbarn och barnsjukhus i Brooklyn, New York. Hon är också en aktiv medlem av Maimonides Medical Center och Kings County Hospital kluven läpp och gommen tvärvetenskapligt Team. Hon har en magisterexamen i vetenskap i genetisk rådgivning från Boston University i Boston, Massachusetts.
1. Sadler TW. Langmans medicinska embryologi. Nionde Upplagan. Sidorna 390-395.
2. Parker SE, Mai CT, Canfield MA, Rickard R, Wang Y, Meyer RE, Anderson P, Mason CA, Collins JS, Kirby RS, Correa A. För det nationella nätverket för förebyggande av fosterskador. Uppdaterade nationella födelseprevalens uppskattningar för utvalda fosterskador i USA. 2004-2006. Fosterskador forskning( Del A): klinisk och molekylär teratologi 2010;88: 1008-1016.
3. Fraser FC. Genetiken av klyftläpp och klyftgom. Är. J. Hum. Genet. 1970;22: 336–352.
4. Han är en av de mest kända. Perikonceptuellt folatintag genom tillskott och matintag minskar risken för icke-syndromisk klyftläpp med eller utan klyftgom. Föregående Med 2004; 39: 689-694.
5. Tan TY. Kilpatrick N, Farlie PG. Utvecklings-och genetiska perspektiv på Pierre Robin sekvens. Är. J. Med. Genet. 2013; 163C: 295-305.
6. McGinn DM, Emanuel BS, Zackai EH. 22q11.2 deletionssyndrom. September. 23, 1999. . I: Pagon RA, Adam MP, Ardinger HH, et al., redaktörer. GeneReviews . Seattle (WA): University of Washington, Seattle; 1993-2014. Tillgänglig från: http://www.ncbi.nlm.nih.gov/books/NBK1523/.
7. Det är en av de mest populära platserna i världen. Wolf-Hirschhorn Syndrom. Apr. 29, 2002. . I: Pagon RA, Adam MP, Ardinger HH, et al., redaktörer. GeneReviews . Seattle (WA): University of Washington, Seattle; 1993-2014. Tillgänglig från: http://www.ncbi.nlm.nih.gov/books/NBK1183/.
8. Robin NH, Moran RT, Ala-Kokko L. Stickler syndrom. Jun. 9, 2000. . I: Pagon RA, Adam MP, Ardinger HH, et al., redaktörer. GeneReviews . Seattle (WA): University of Washington, Seattle; 1993-2014. Tillgänglig från: http://www.ncbi.nlm.nih.gov/books/NBK1302/.
9. Katsanis SH, Jabs EW. Treacher Collins Syndrom. Jul. 20, 2004. . I: Pagon RA, Adam MP, Ardinger HH, et al., redaktörer. GeneReviews . Seattle (WA): University of Washington, Seattle; 1993-2014. Tillgänglig från: http://www.ncbi.nlm.nih.gov/books/NBK1532/.
10. Schutte BC, Saal HM, Goudy S, et al. IRF6-relaterade störningar. Okt. 30, 2003. . I: Pagon RA, Adam MP, Ardinger HH, et al., redaktörer. GeneReviews . Seattle (WA): University of Washington, Seattle; 1993-2014. Tillgänglig från: http://www.ncbi.nlm.nih.gov/books/NBK1407/.