gränser i pediatrik
- introduktion
- Långt QT-syndrom
- Lqts1
- LQTS2
- LQTS3
- LQTS4
- LQTS5
- LQTS6
- LQTS7
- LQTS8
- LQTS9 och LQTS10
- LQTS11
- LQTS12
- LQTS13
- förvärvade LQTS
- elektrokardiografiska egenskaper i de tre vanliga formerna av långt QT-syndrom
- genotyp-fenotyp
- klinisk hantering av LQTS
- LQTS: Saudi perspektiv
- intressekonflikt uttalande
- bekräftelser
introduktion
familjära eller ärftliga hjärtarytmier omfattar betydande procentandelar av arytmier och även orsak till plötslig hjärtdöd (SCD) (1, 2). Under de senaste två decennierna har forskare och kliniker gett enorma ansträngningar för att riva upp de invecklade och komplexa mekanismerna för medfödda familjära arytmier (1-8). För att förstå mekanismen för arytmogenes behöver vi veta grunderna för hjärtcellstruktur och deras elektrofysiologiska egenskaper. Hjärtmyocyter är de viktigaste fungerande cellerna i hjärtat och de är i stor utsträckning kopplade så att impulser sprids snabbt och enhetligt. Kardiomyocyter separeras från varandra genom en specialiserad gräns som kallas den interkalerade skivan; gap-korsningsproteiner, hjärt-desmosomer och jonkanaler finns i den interkalerade skivan (9). Gap korsningar består av tätt packade connexiner som tillåter intercellulär utbyte av små molekyler och även tillåta att strömma excitatoriska strömmar från en cell till dess angränsande cell. Desmosomer tillsammans med adherens-korsningarna är ansvariga för de mekaniska bindningarna hos enskilda kardiomyocyter. Alla dessa komponenter i de interkalerade skivorna är sekventiellt segregerade och varje komponent utövar sin unika funktion; störning av den ena komponenten påverkar funktionen hos andra komponenter, vilket predisponerar hjärtat för att utveckla arytmier (9-11). Hjärtjonkanaler är porbildande proteinkomplex som ger spänningsgrindade och intrikat samordnade inre och yttre rörelser av joniska strömmar över cellmembranen, väsentliga för hjärtrytmgenerering och förökning. Långt QT-syndrom (LQTS), kort QT-syndrom (SQTS), sjukt sinus syndrom (SSS), hjärtledningsdefekt (CCD), BrS, katekolaminerg polymorf ventrikulär takykardi (CPVT), tidigt repolariseringssyndrom (ERS) och familjär förmaksflimmer (AF) är de för närvarande kända hjärtkanalopatierna, som kan uppstå på grund av en enda eller multipel defekt i generna kopplade till hjärtrytmgenerering och förökning.
i den här översynen kommer vi att beskriva uteslutande på LQTS-kopplade arytmier, dess patofysiologi och för närvarande tillgänglig klinisk hantering. Vi har banat väg för att belysa genetisk patologi i familjär hjärtarytmier i Saudiarabien (1, 12-14), Vi genomför fortfarande kardiogenetiska undersökningar i Saudiarabien, i slutet av denna översyn kommer vi att diskutera den för närvarande tillgängliga kunskapen om de genetiska och kliniska fynd som erhållits från flera saudiarabiska familjer med historia av synkope och SCD. Vi kommer också att lägga till de nyligen publicerade uppgifterna om LQTS från ett team vid King Faisal Specialist Hospital and Research Center, Riyadh (15).
Långt QT-syndrom
medfödd LQTS är en ärftlig störning definierad genom förlängning av QT-intervallet på elektrokardiogram (EKG). Patienter med alla former av LQTS är predisponerade för ventrikulär takyarytmi, torsades de pointes (TdP) som leder till återkommande synkope eller SCD. I många fall kan synkope eller plötslig död vara den första och den enda manifestationen. LQTS påverkar uppskattningsvis 1 av 2 000 personer över hela världen (16). Kännetecknet för LQTS är förlängning av QT-intervallet på EKG (korrigerat för hjärtfrekvens, dvs QTc). Normala värden för QTc är 440 ms hos män och 450 ms hos kvinnor. Hos barn är ålders-och könsberoende värden relevanta. Senaste konsensusrekommendation för LQTS-diagnos är följande (17, 18):
1. LQTS diagnostiseras:
a. i närvaro av en LQTS-riskpoäng 3,5 i frånvaro av en sekundär orsak till QT-förlängning och/eller
b. i närvaro av en otvetydigt patogen mutation i en av LQTS-generna eller
c. I närvaro av ett QT-intervall korrigerat för hjärtfrekvens med Bazetts formel (QTc) 500 ms i upprepat 12-bly elektrokardiogram och i frånvaro av en sekundär orsak till QT-förlängning.
2. LQTS kan diagnostiseras i närvaro av en QTc mellan 480 och 499 ms i upprepade 12-bly EKG hos en patient med oförklarlig synkope i frånvaro av en sekundär orsak till QT-förlängning och i frånvaro av en patogen mutation.
patienter med QTc-varaktighet 500 ms hade en signifikant högre kumulativ sannolikhet att uppleva sitt första synkope jämfört med patienter med QTc-varaktighet <500 ms från födseln till 20 års ålder (19). Men patienter som har upplevt 2, 3 och 4 synkopepisoder var risken för en efterföljande synkopepisod nästan identisk mellan patienter som hade smal eller långvarig QTc-varaktighet (19). Den molekylära grunden för LQTS är heterogen och hittills har mutationer i 13 olika gener beskrivits kausala till LQTS (1-8, 19, 20). En genetisk defekt finns vanligtvis hos 70% av LQTS-patienterna i en av dessa 13 gener (1-8, 19, 20). Bland de för närvarande kända 13 olika typerna av LQTS är de vanligaste LQTS1, LQTS2 och LQTS3 på grund av defekter i hjärtjonkanalgener, KCNQ1, KCNH2 respektive SCN5A. Nittio procent av alla LQTS kausala mutationer finns i dessa tre gener (1-8, 19, 20). Mutationer i de återstående 10 generna är sällsynta och omfattar endast 10% av alla de för närvarande kända LQTS-mutationerna.
Långt QT-syndrom är vanligtvis en autosomal dominerande sjukdom, men ibland kunde flera mutationer i en enda gen eller i olika gener hittas hos 5-10% av patienterna med LQTS (21, 22). Patienter med multipla mutationer kan uppvisa en längre QTc jämfört med de med en enda mutation och sådana patienter har också 3,5 gånger ökad risk för livshotande hjärthändelser (21, 22)
Lqts1
mutationer i kcnq1-genen är den vanligaste formen av alla LQTS och kallas LQTS typ 1 (LQTS1). Femtio procent av alla LQTS kausala mutationer finns i kcnq1-genen (20). KvLQT1 (även kallad Kv7. 1) är ett protein tillverkat av kcnq1-genen och proteinet bildar tetramerer i endoplasmatisk retikulum inuti cellerna, som förmodas sammonteras med proteinet minK (kodat av KCNE1) och transporteras sedan till plasmamembranet i hjärtmyocyterna, där de förmedlar en långsamt aktiverande ström som accelererar repolariseringen av åtgärdspotentialen i hjärtvävnader, denna ström är känd som IKs (Figur 1). Lqts1 kausala kcnq1 mutationer är mestadels missense mutationer och i sällsynta fall kan de vara frameshift mutationer i C-terminala regionen (23). Hjärtarytmi i kcnq1 mutationsbärare utlöses av adrenerga enheter, t ex emotionell stress, fysisk ansträngning, dykning, simning (24-26). Patienter med mutationer vid transmembrandomänerna i KvLQT1 har högre risk för LQTS-relaterade hjärthändelser och har större känslighet för sympatisk stimulering (26, 27). Patienter med missense mutationer löper ökad risk jämfört med patienter med icke-sense eller trunkerande mutationer (27, 28). Variation i 3′-UTR i kcnq1-genen påverkar också arytmikänsligheten signifikant, förmodligen genom att påverka genuttrycket (29). Patienter med arytmier på grund av kcnq1-mutationer svarar ganska bra på hCG-blockerare, men vissa patienter kan fortfarande vara mindre mottagliga eller till och med resistenta mot detta läkemedel. I en ny artikel, Barsheshet et al. (30) hävdade att patienterna med mutationer utanför cytoplasmatisk slinga (c-slinga)-regionen i KvLQT1 är mindre lyhörda för hCG-blockerare.
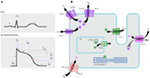
Figur 1. Joniska strömmar som bidrar till ventrikulär aktionspotential (A) och schematisk representation av en kardiomyocyt som visar (endast) de proteiner som är involverade i patogenesen av ärftliga arytmisyndrom (B). I (A) är åtgärdspotentialen anpassad till dess ungefärliga verkningstid under EKG. I (B) är ankyrin-B, ett adapterprotein involverat i det långa QT-syndromet typ 4, inte avbildat.
homozygota eller sammansatta heterozygota mutationer i kcnq1-genen, som kan orsaka den recessiva formen av sjukdomen, Jervell och Lange-Nielsen syndrom (JLNS), typ 1 (31), är ganska sällsynta. Jlns-patienter lider också av allvarliga hjärtarytmier och dövhet (31, 32). Patienter med kcnq1-mutationer som orsakas av JLNS har vanligtvis inga funktionella IKs (28, 31-33). Det är möjligt att vissa patienter inte kan ha någon dövhet trots att de har homozygota eller sammansatta heterozygota mutationer i KCNQ1, kallas sådana fall autosomal recessiv LQTS1 (13, 32). Hos dessa patienter kan en liten mängd funktionell iks-ström (<10% av den totala IKs) fortfarande vara närvarande, vilket upprätthåller hörselfunktionen, men deras hjärtrytmfel är lika allvarliga som hos JLNS-patienter (13, 32).
LQTS2
denna typ av LQTS är lika vanlig som LQTS1 och står för 35-40% av LQTS-patienter med en detekterbar mutation (1, 20, 24, 25). KCNH2 kodar för HERG-proteinet (Kv11.1), vilket är subenheten för den snabbt aktiverande fördröjda likriktaren k+ current (IKr). Patogena mutationer i denna gen som minskar Kv11.1-kanalfunktionen förlänger QT-intervallets varaktighet (Figur 1) och är orsakssamband med LQTS2. Tjugonio procent av synkopalattackerna i LQTS2 inträffar under vila / sömn och endast 13% av synkopalattackerna rapporterades inträffa under träning (25, 34). Plötsliga häpnadsväckande ljud, t.ex. väckarklockor, dörrklockor, telefonringar, utlöser vanligtvis syncopala attacker hos dessa patienter (24, 34). Patienter med mutationer i den porbildande regionen i Kv11.1 (kodad av kcnh2-genen) är mottagliga för hög risk för arytmirelaterade hjärthändelser jämfört med patienter med icke-porregionmutationer (35). Hos nyfödda är 2: 1 atrioventrikulärt (AV) block företrädesvis associerat med KCNH2-mutationer (36). Komplett AV-block komplicerat av LQTS hittades också hos 17% av vuxna patienter med en mutation i kcnh2-genen (37). Homozygota mutationer i KCNH2 är sällsynta och när de är närvarande lider patienter av en allvarlig form av LQTS, med 2:1 av-block och svåra ventrikulära arytmier, under intrauterina stadier såväl som efter födseln (11, 38-40). Förutom de många mutationer som beskrivs i lqts2-patologi kan vanliga polymorfa varianter i kcnh2-genen också modulera sjukdomens svårighetsgrad. Ett spännande exempel är K897T polymorfism (SNP) i KCNH2, som är närvarande i 33% av den allmänna befolkningen (41). K897T rapporterades förvärra patogeniciteten hos kcnh2-mutationerna (42, 43).
LQTS3
Nav1.5 är den porbildande subenheten av den porbildande subenheten av den spänningsberoende hjärt-na+-kanalen, det är ett integrerat membranprotein kodat av SCN5A-genen och är involverat i initiering och ledning av hjärtverkningspotentialer. Cardiac Na + – kanaler består av en porbildande Bisexuell-underenhet (kodad av SCN5A) och en eller flera hjälp-underenheter av typen.
LQTS3 orsakas av förstärkning av funktionsmutationer som stör den snabba inaktiveringen av subenheten hos den subenheten (Figur 1) och en mutation i scn5a-genen har beskrivits hos <10% av alla LQTS-patienter med en mutation (1, 20, 24). Lqts3-patienter upplever majoriteten (39%) av sina hjärthändelser under sömn/vila (25), och cirka 13% av händelserna rapporterades inträffa under träning (25). I flera fall visades en enda SCN5A-mutation utöva två eller till och med tre distinkta fenotyper av arytmier i samma familj, t.ex. LQTS, BrS eller CCD (44-47). Manliga patienter med en lqts3-mutation kan utveckla symtom mycket tidigare än kvinnliga patienter (48). Både heterozygota och homozygota SCN5A-mutationer har beskrivits i LQTS3 med funktionellt 2: 1 av-block (49). Vi har nyligen hittat en iransk Familj med 1507_1509delqkp-mutationen hos flera familjemedlemmar, där patienter hade kombinerat LQTS och CCD (data visas inte). Denna mutation har också rapporterats hos patienter från andra länder, vilket tyder på att detta är en återkommande och hot spot-mutation . Mutationer med en sådan förlust-och förstärkningsfunktionsegenskaper under olika faser av åtgärdspotentialen rapporterades också i 1493delk och 1795insd-mutationer (44, 51).
ibland kan en SNP också utöva patogen effekt på dess bärare. S1103Y är en vanlig variant i SCN5A-genen, närvarande i 13% av afroamerikanerna (52). Bärare med denna variant har ökad risk för arytmier och plötsligt spädbarnsdödssyndrom (SIDS) (52).
LQTS4
lqts4 representerar den första icke-kanalformen av LQTS. En mutation i ANK2, ett adapterprotein, leder till intracellulär kalciumöverbelastning som bidrar till LQTS4 (53, 54). Förutom QT-förlängning kan patienter med detta syndrom ha sinus bradykardi, paroxysmal AF och CPVT (54). Patogen effekt av ANK2-mutationerna kan vara måttlig till svår och de kliniska uttrycken beror på mutationens svårighetsgrad.
LQTS5
mutationer i KCNE1 är associerade med LQTS5 (Figur 1) (55, 56). Heterozygota kcne1-mutationer minskar IKs genom att utöva en dominerande negativ effekt på dess åtföljande normala allel och leda till försenad hjärtrepolarisering (Figur 1), ansvarig för ökad risk för arytmier (6). Patienter med homozygota kcne1-mutationer lider av JLNS (typ 2) (57, 58).
D85N är en polymorfism i kcne1-genen, närvarande i 0.7-1% av den allmänna befolkningen (41). I en studie av Nishio et al. (59), d85n-polymorfism hittades oftare hos LQTS-patienterna, vilket gjorde det till en riskgenotyp i LQTS-patologi (potentiellt endast i den asiatiska befolkningen). I Europa rapporterades D85N hos 5% av förvärvade LQTS (aLQTS) patienter (i en kohort på 32 patienter), som hade upplevt TdP (60).
LQTS6
kcne2-genen kodar för minkrelaterad peptid 1 (MiRP1), en förmodad subenhet av den kardiovaskulära kaliumkanalen IKr (Figur 1). Mutationer i kcne2-genen kan också leda till defekter i den snabbt aktiverande komponenten i den fördröjda likriktarens kaliumström (IKr), patologisk grund för LQTS6 (61). Auditiv / akustisk stimulans som väckarklocka buller, dörrklocka etc. kan provocera syncopala attacker i kcne2-mutationsbärare, liknande KCNH2-mutation (62).
LQTS7
detta syndrom är också känt som Andersen–Tawil syndrom (ATS). ATS är en sällsynt sjukdom, manifesterad av tillfällig synkope och hjärtstillestånd. EKG-funktioner inkluderar mild Qt-intervallförlängning, onormala U-vågor, frekvent ventrikulär ektopi, dubbelriktad ventrikulär takykardi (VT) och polymorf VT. Detta syndrom uppvisar också extrakardiella egenskaper, t.ex. skelettmuskulatur periodisk förlamning och utvecklingsproblem, såsom klyftgom, låga öron, kort statur och utvecklingsfel i lemmarna (63). Majoriteten av kliniskt diagnostiserade ATS-patienter rapporterades ha en mutation i KCNJ2 (63). KCNJ2 kodar för en porbildande underenhet av inåt likriktande kaliumkanaler (IK1) (Figur 1) (64, 65).
LQTS8
även känd som Timothy syndrom (TS), patienter visar svår QT-förlängning på sina EKG, som kombineras med syndaktyli, skallighet vid födseln, och små tänder i 100% av fallen och mindre penetrerande hjärt strukturella missbildningar, mental retardation, autism, och ansikts dysmorfa funktioner (66). Det finns två undertyper: TS1 (klassisk) och TS2 (sällsynt form).
TS2 är kardiologiskt strängare än TS1 (66, 67). TS2-patienter saknar också syndaktiskt (67). Mutationer i subenheten av den l-typ av kalciumström (ICA-L) som kodar för genen CACNA1C leder till båda formerna av TS (LQTS8). Det finns alternativt skarvade, ömsesidigt uteslutande exon – 8 i CACNA1C-genen, för tydlighetens skull, de heter som exon-8 och exon-8A.i hjärta och hjärna, där CACNA1C huvudsakligen uttrycks, exon-8 finns i 80% av mRNA-transkripten och exon-8A är närvarande 20% transkriptioner (66). G406r-mutation i exon-8A är kausal till klassisk form av TS (TS1) och G402S i exon-8 rapporterades i svårare i TS2. Ett nytt tillägg till denna lista är Ala1473Gly-mutation, som har beskrivits i ett TS-spädbarn med ytterligare expanderad fenotyp (68). Alla mutationer var antingen de novo eller mosaik i föräldern och all resultatförstärkning av funktionen hos ICA-L-kanalen (66-69).
LQTS9 och LQTS10
mutationer i CAV3 eller SCN4B ger förstärkning av funktion i sen INa, vilket orsakar en lqts3-liknande fenotyp (70-74). De är kända som LQTS9 (associerad med CAV3-mutation) och LQTS10 (associerad med SCN4B-mutation).
Caveolae beskrivs snyggt av Engelman et al. (72) som “små grottor” i plasmamembranet. De är små obelagda gropar och anses vara platsen för viktiga dynamiska och reglerande händelser vid plasmamembranet (72, 73). Caveoliner är de viktigaste proteinerna i caveolae, och caveolin-3 (kodad av Gen CAV3) finns specifikt i kardiomyocyter och skelettmuskelceller. Flera hjärtjonkanaler har specifikt rapporterats vara lokaliserade i caveolae extraherade från hjärtmyocyter som berikas i caveolin-3 (72, 73). Dessutom är komponenter i den signaleringskaskaden för den 2-adrenerga receptorn också närvarande i caveolae-berikade membran (72, 73).
SCN4B kodar för nav Macau4, som är en hjälp-underenhet av hjärtnatriumkanalen. Hittills har endast en mutation (L179F) rapporterats i denna gen i en mexikansk familj med flera drabbade familjemedlemmar (74). Mutation i denna gen befanns resultera i förstärkning av funktionen hos Nav1.5-strömmen (74).
LQTS11
i hjärtat medieras sympatisk reglering av hjärtverkningspotentialvaraktighet (APD) genom aktivering av actui-adrenerg receptor (actui-ar), vilket kräver montering av AKAP9 (Yotiao) med iks-kanalens subenhet (KvLQT1). Mutation i AKAP9 orsakar LQTS11 (75). Hittills har endast en mutation, S1570L, i AKAP9 rapporterats (75).
LQTS12
-Mutation i genen för chubbi-1-Syntrofin (SNTN1) är orsakssamband med lqts12. Mutation i denna gen leder till förstärkning av funktionen hos hjärtnatriumkanalen (Nav1.5), som är den patologiska grunden för LQTS12 (76).
LQTS13
g proteinkopplad, inåt likriktande kaliumkanalsubenhet (Kir3.4) kodas av kcnj5-genen. En förlust av funktionsmutation i denna gen kan orsaka LQTS13 (77). Hittills har endast en mutation, G387R, beskrivits i en kinesisk familj med nio patienter som har denna mutation. Reducerat plasmamembranuttryck av Kir3.4 föreslogs som patologi för LQTS hos patienterna.
förvärvade LQTS
förutom de medfödda LQTS finns också en annan variant av LQTS som kallas aLQTS, vilket orsakas av faktorer och ämnen som minskar kaliumflödet och försämrar myokardiums förmåga att repolarisera. Välkända tillstånd är kvinnligt kön, hypokalemi och läkemedel som hämmar hjärtkaliumkanaler (78-80). Ett antal vanligt föreskrivna läkemedel kan också företrädesvis binda och blockera HERG-kanalen (Kv11.1, ett protein kodat av kcnh2-genen) och predisponerar för aLQTS (79, 80). Nyligen visades det att blockad av IKs också kunde bidra till läkemedelsinducerad aLQTS, speciellt när repolariseringsreserven äventyras (81). Fluoxetin och norfluoxetin befanns undertrycka iks-egenskaperna, både in vivo och in vitro och ledde till markerade LQTS (81). Polymorfismer, D85N i minK (gen KCNE1), T8A, Q9E i MiRP1 (gen KCNE2), som är förmodade underenheter av iks-och IKr-kanalerna, rapporterades orsaka aLQTS (82, 83). Autoimmuna LQTS har också rapporterats hos en patient med IgG innehållande Anti-HERG-antikroppar (84).
elektrokardiografiska egenskaper i de tre vanliga formerna av långt QT-syndrom
typiska ST-t-vågmönster finns i majoriteten av genotypade LQTS-patienter och kan användas för att identifiera lqts1, LQTS2 och eventuellt lqts3-genotyper (85, 86).
lqts1-formen av LQTS är associerad med en bred T-våg utan förkortning av QT-intervallet vid träning (figur 2a). LQTS2 är associerad med låg amplitud, ofta bifid, T-vågor (Figur 2B). LQTS3 är förknippat med ett långt iso-elektriskt segment och en smalbaserad, lång T-våg (figur 2C). Pausberoende av TDP-debut hos medfödda LQTS är genotypspecifikt och är dominerande i LQTS2 men nästan frånvarande i LQTS1 (87).

Figur 2. Elektrokardiograminspelningar från patienter med LQTS1, LQTS2 och LQTS3. (A) 12-avlednings-EKG hos en 18-årig man med en kcnq1-mutation. QT-intervallet förlängs(QTc = 500 ms. ST-segmentet har en bred bas och relativt stor amplitud. Ledningsintervallet är normalt (standardkalibrering). (B) 12-avlednings-EKG hos en 14-årig flicka med en kcnh2-mutation. QT-intervallet förlängs(QTc 520 ms). ST-segmentet är hakat i bly V3 och har relativt låg amplitud i extremitetsledningarna. Ledningsintervallet är normalt (standardkalibrering). (C) 12-bly-EKG hos en 12-årig pojke med en SCN5A-mutation. QT-intervallet förlängs(QTc 600 ms). ST-segmentet har ett långt (nästan) iso-elektriskt segment med en stor, skarp och smal T-våg. Ledningsintervallet är normalt (standardkalibrering).
även om mönster kan föreslå en specifik genotyp av LQTS, har undantag ofta hittats.
genotyp-fenotyp
Långt QT-syndrom är en autosomal dominant sjukdom. Genotyp-och fenotypanalys bland de heterozygota mutationsbärarna har genomförts ganska omfattande hos lqts1 -, LQTS2-och LQTS3-patienterna. Under barndomen är risken för hjärthändelser signifikant högre hos lqts1-män än hos lqts1-kvinnor, medan inga signifikanta könsrelaterade skillnader i risk för hjärthändelser observerades bland lqts2-och LQTS3-patienter (88-91). Under vuxen ålder (även efter 40 års ålder) kan lqts1-och LQTS2-kvinnor ha signifikant högre risk för hjärthändelser än respektive män (88-91). I allmänhet verkar dödlighet av hjärthändelser vara dominerande hos lqts3-patienter än hos lqts1-och LQTS2-patienter (91). Kvinnor med LQTS har en minskad risk för hjärthändelser under graviditeten, men risken ökar ganska under 9-månaders postpartumperioden, speciellt hos kvinnor med mutation i kcnh2-genen (92).
plötslig hjärtdöd hos barn kan också orsakas av mutationer i hjärtjonkanalgenerna (93-96). Cirka 28% av barnen med en oförklarlig SCD (mellan 1 och 18 år, medelålder: 12,3 3,8 år) var bärare av mutationer i LQTS kausala gener (97). I SIDS verkade mutationer i SCN5A dominerande (98, 99), men mutationer i KCNQ1, KCNH2, KCNE2 och CAV3, SCN4B och SCN3B hittades också (100, 101). Intrauterin fosterdöd rapporterades också på grund av defekter i hjärtjonkanalgener (12 102).
klinisk hantering av LQTS
upphörande av alla läkemedel som är kända för att förlänga QT-intervallet och även korrigering av elektrolytobalanser och/eller utfällande metaboliska tillstånd bör vara det primära fokuset vid behandling av (förvärvade) LQTS-patienter. Symtom i LQTS är ofta adrenergiskt medierad, därför rekommenderas vanligtvis begränsning av patienternas deltagande i atletiska aktiviteter (24, 25). Stöttepelaren i klinisk terapi för LQTS är AUC-blockad. Långverkande preparat av propranolol, nadolol och metoprolol används vanligtvis, och deras effektivitet vid XXL-blockad bedöms genom avtrubbning av träningspulsen (t ex genom >20%) (24, 25). Bland alla de oc-blockerare, propranolol och nadolol anses överlägsen än metoprolol hos symptomatiska patienter (103). Vidare kan även användas som profylaktisk behandling i tysta mutationsbärare för att minska SCD (24). I en studie av Barsheshet et al. (30), patienter med C-loop missense-mutationer i kcnq1-genen uppvisade hög risk för livshotande hjärthändelser och de hade betydande fördelar med behandlingen med HCG-blockerare. Eftersom kvinnor med LQTS2 har en ökad risk under 9-månaders postpartumperioden, bör ovariablockerare ordineras för att minska eventuella hjärthändelser under denna högriskperiod (92). En implanterbar kardioverter-defibrillatorer (ICD) kan övervägas för patienter med återkommande synkope trots behandling med en blockerare eller hos patienter med hög risk för hjärtstillestånd (t. ex. lqts2 och LQTS3 med en dokumenterad QTc-förlängning). Vänster hjärt sympatisk denervering (LCSD) rekommenderas för högrisk LQTS-patienter i vilka en ICD är kontraindicerad eller nekad och HCG-blockerare är antingen inte effektiva, inte tolererade, inte accepterade eller kontraindicerade (18, 104). Jlns-patienter har vanligtvis en QTc >500 ms och har också hög risk.
LQTS: Saudi perspektiv
första rapporten om LQTS från Saudiarabien publicerades 1993 från Riyadh Armed Forces Hospital (105). Fyra spädbarn och småbarn mellan 6 och 48 månader med historia av återkommande anfall, från en enda familj, diagnostiserades som LQTS (105). Familjehistoria avslöjade att två andra utökade familjemedlemmar hade liknande episoder av plötslig medvetslöshet och tre familjemedlemmar dog plötsligt (105). I samtliga fall var initial diagnos epilepsi (105). Flera år senare har två sporadiska fallrapporter med jämförelsevis svårare variant av neonatal LQTS kombinerat med 2:1 av-block rapporterats (106, 107). Alla dessa publicerade kliniska rapporter var utan några genetiska fynd som kunde förklara patofysiologin för LQTS i dem (105-107).
vi har för första gången rapporterat genetiska defekter som en patologisk grund för LQTS hos liknande patienter från Saudiarabien. Vi har undersökt sex-saudiska familjer med synkopehistoria och plötsliga oförklarliga dödsfall hos foster, nyfödda och barn (12-14). Autosomal recessiv LQTS1 diagnostiserades hos barn från två familjer (Figur 3). Autosomal recessiv lqts2 diagnostiserades i två familjer (Figur 4). I en familj diagnostiserades en kvinnlig patient med autosomal dominant LQTS2 (Figur 5), patienten hade synkopeattacker under återhämtningsperioden efter förlossningen på sjukhuset, vilket är mycket vanligt hos KCNH2-mutationsbärare kvinnor. I alla våra patienter ledde identifiering av patogena mutationer i LQTS kausala hjärtjonkanalgener till en bekräftad klinisk diagnos, som felaktigt diagnostiserades som epileptiska kramper innan de hänvisades till oss (13, 14) nyligen, Shinwari et al. (15) från King Faisal Specialist Hospital and Research Center rapporterade en lqts1 kausal KCNQ1-mutation, H258P, i en stor familj med 12 drabbade individer. Endast två bärare var symptomatiska, deras QTc var >500 ms, och sackaros-blockerare undertryckte de kliniska symtomen hos en patient och den andra symptomatiska patienten krävde en ICD.
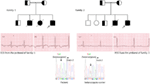
Figur 3. Stamtavla av familjen-1 och 2 med autosomal recessiv LQTS1, probands visas med en pil. EKG från probanderna i två familjer visas i mitten, med en QTc på 557 respektive 529 ms. Intronisk mutation, c. 387 -5 T > A i kcnq1-genen hittades hos patienterna (visas i botten). Mutation visades med en pil. Icke-fyllda cirklar och kvadrater är icke-bärare för mutationen. Berörda individer visas som fyllda cirklar (kvinnliga) och rutor (manliga). Halvfyllda rutor och cirklar är individer med heterozygotmutation. Avlidna individer indikeras med snedstreck, probander indikeras med en pil och konsanguineous äktenskap indikeras med = Exon − intron gräns visas med en prickad linje med pil som pekar mot exon.
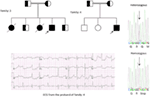
Figur 4. Stamtavla ritning av familjen-3 och 4 med autosomal recessiv LQTS2. EKG av proband från family-4 visas under stamtavlan, som visar sinus takykardi, nästan fullständigt AV-block, bred komplex flyktrytm med mycket långa QTc-intervaller (QT >600 ms). Mutation, c.3208 C > T (p.Q1070X) i kcnh2-genen hittades hos patienterna (visas till höger), markerad med en pil.
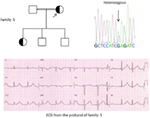
Figur 5. Överst till vänster: stamtavla i familjen-5. Berörd individ visas av fylld cirkel (kvinnlig) Proband indikeras med en pil. 12-bly EKG av proband (I:2). EKG visar en hög topp, bred baserad, stor amplitud T-våg, QTc-intervallet är 580 ms. Screening av kcnh2-genen visar substitution av nukleotid “G” för en ” A “(c.2362g > a, pil markerad), vilket leder till aminosyrasubstitution, s. E788K.
genetiska och kliniska fynd i vår studie (12-14) från Saudiarabien är ganska spännande av flera anledningar: (1) Totalt har vi undersökt sexfamiljer, bland dem var fyra homozygota / sammansatta heterozygota för mutationerna och mutationerna härstammar från en förfäderkälla; (2) alla mutationer i LQTS var nya, rapporterade endast i dessa Arabiska familjer; (3) på grund av homozygositeten eller sammansatta heterozygositeten för mutationerna var kliniska fenotyper också allvarliga i våra studerade familjer (12-14). Vi föreslog att de genetiska och fenotypiska observationerna härrörde från den extremt höga frekvensen av äktenskapliga äktenskap i Saudiarabien (108, 109). Vår studie gav de första vetenskapliga bevisen om konsanguinitetens roll i att utöva en central roll i oförklarliga hjärtarytmier och SCDs hos barn och ungdomar i Saudiarabien (12-14). Vi har också visat att kcnq1-mutationen c. 387 -5 T > a (NM_000218) (Figur 3) hade spridit sig i Assir-provinsen Saudiarabien från en gemensam förfader under flera generationer på grund av hög förekomst av blodsförvanter äktenskap . Hittills är detta den vanligaste lqts1-kausalmutationen i Saudiarabisk befolkning, som också observerades vid King Faisal Specialist Hospital och vid Khamis Mashayt Military Hospital (opublicerat). Ett liknande resultat erhölls också för lqts2 kausal mutation, c.3208 C > T (p.Q1070X) (Figur 4) i kcnh2-genen (12, 14), som också är en grundmutation i Saudiarabien och segregerad under många generationer i Assir-regionen (se kartan, Figur 6). Vi skulle förutsäga att det finns ett stort antal individer med de nämnda förfäderna/grundmutationerna (både i KCNQ1 och KCNH2 och andra gener) i denna region och även i de stora städerna som Riyadh, Jeddah och Dammam på grund av urban migration. Mer grundare eller förfäders mutationer patogena till LQTS är mycket förutses i andra provinser i Saudiarabien på grund av hög grad av blodsförvanter äktenskap.
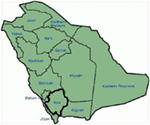
Figur 6. Karta över Saudiarabien (med tillstånd: Wikipedia). Assir-regionen är markerad med tjock linje, där vi har hittat grundmutationerna i kcnq1-och KCNH2-gener, beskrivna i familj 1-4.
eftersom LQTS är en autosomal dominant sjukdom, förväntade vi oss att observera övervägande heterozygota mutationsbärarpatienter. Men i vår studie identifierade vi främst patienter med recessiva mutationer (12-14). Klart, dessa patienter blir första symtomatiska och, hänvisades huvudsakligen till Prince Sultan Cardiac Center, föra dem under vår uppmärksamhet. Ändå tyder resultaten från våra undersökningar på att recessiva LQTS kan ha dödliga kliniska fenotyper hos barn, och de är inte ovanliga i Saudiarabien (12-14). Eftersom de heterozygota mutationsbärarna också är mottagliga för att utveckla arytmi och dess komplikationer, bör samordnade initiativ tas för att få de lokala allmänläkarna, kardiologerna och kliniska genetikerna i en gemensam plattform för att identifiera individerna i riskzonen. Vidare har vi för närvarande ingen genetisk information för andra familjära arytmistörningar, t.ex. CPVT, SQTS, BrS, AF, etc. Specialiserade kardiogenetiska centra bör ta initiativ till att söka efter genetiska defekter, mutationer och utföra genotyp-fenotypstudier i alla former av ärftliga arytmier. Det bör också hållas i beaktande att inte alla genetiska arytmier skulle ha en familjehistoria, som i många fall arytmi kausal mutation är de novo ursprung, dvs proband är den första patienten i den familjen med mutationen och han/hon är källan för att överföra mutationen i nedströms generationer (110, 111). På grund av den mycket höga andelen blodsförvanter äktenskap i Saudiarabien, vi förväntar oss mycket mer grundare mutationer utövar en avgörande roll i medfödda arytmier i detta land. Identifiera dessa grundare mutationer bör vara vår första och främsta uppgift, vilket skulle underlätta för oss att utveckla en effektiv premarital och även pre-symtomatisk genetisk rådgivning i detta land. Mutationer i LQTS kausala gener kan ge variation i klinisk penetrans, i en extrem kan vissa bärare förmodligen vara helt friska, men vissa bärare kan ha sin första manifestation av sjukdomen som synkope eller plötslig död. Plötslig död hos ett ungt barn eller en vuxen utgör en stor psykologisk och känslomässig börda för familjen, screening av bärarna är väsentliga eftersom det finns enkel medicinering, t.ex. Individer med homozygota mutationer i LQTS kausala gener kan ha svår sjuklighet och extremt hög dödlighet inklusive fetala brady-takyarytmier, och i många fall missfall, spontana aborter hos gravida mödrar (12-14).
inte mycket forskning har också genomförts i Saudiarabien om förekomsten av olika SNP i de arytmibundna generna och även i generna som reglerar dem. Splawski et al. (112) beskrev en vanlig s1103y-variant i SCN5A-genen associerad med arytmi hos afroamerikaner. Variantallelen benämnd Y1103 är ansvarig för att accelerera kanalaktivering, vilket ökar sannolikheten för hjärtarytmier hos personer med afrikansk härkomst (52, 112). K393N är en variant i kcnq1-genen som rapporterats hos lqts1-patienter i USA, men i den arabiska befolkningen har vi upptäckt denna variant hos 2% av individerna (opublicerade data). Huruvida k393n-varianten i kcnq1-genen i araber är jämförbar med s1103y (SCN5A)-varianten i afroamerikaner eller d85n (KCNE1) – varianten i den japanska befolkningen kan också förtjäna undersökning (59).
intressekonflikt uttalande
författarna förklarar att forskningen genomfördes i avsaknad av kommersiella eller finansiella relationer som kan tolkas som en potentiell intressekonflikt.
bekräftelser
vår uppriktiga tacksamhet till Prof.Arthur Wilde, Prof. Connie Bezzina, och utgivaren av tidskriften “Heart” för deras tillstånd att använda Figur 1 i detta manuskript från Ref. (113).
1. Bhuiyan ZA. Kliniskt och genetiskt spektrum av ärftliga Hjärtarytmisyndrom. Amsterdam: Amsterdams Universitet (2009).
2. Priori SG, Aliot E, B. L. C., Bossaert L, Breithardt G, Brugada P, et al. Arbetsgruppen för plötslig hjärtdöd, European society of cardiology. Europace (2002) 4:3-18. doi: 10.1053 / eupc.2001.0214
CrossRef Fullständig Text
3. Wang Q, Shen J, Splawski I, Atkinson D, Li Z, Robinson JL, et al. Scn5a-mutationer associerade med en ärftlig hjärtarytmi, långt QT-syndrom. Cell (1995) 80:805-11. doi:10.1016/0092-8674(95)90359-3
Pubmed Abstrakt / Pubmed fulltext / CrossRef fulltext
4. Jag är en av de mest populära i världen. En molekylär grund för hjärtarytmi: HERG-mutationer orsakar långt QT-syndrom. Cell (1995) 80:795-803. doi: 10.1016/0092-8674(95)90358-5
Pubmed Abstrakt / Pubmed Fulltext / CrossRef Fulltext
5. Wang Q, Curran mig, Splawski I, bränna TC, Millholland JM, VanRaay TJ, et al. Positionell kloning av en ny kaliumkanalgen: KVLQT1-mutationer orsakar hjärtarytmier. Nat Genet (1996) 12:17-23. doi: 10.1038 / ng0196-17
PubMed Abstrakt / Pubmed fulltext / CrossRef fulltext
6. Splawski I, Tristani-Firouzi M, Lehmann MH, Sanguinetti MC, Keating MT. Mutationer i hmink-genen orsakar långt QT-syndrom och undertrycker IKs-funktionen. Nat Genet (1997) 17:338-40. doi: 10.1038 / ng1197-338
Pubmed Abstrakt / Pubmed fulltext / CrossRef fulltext
7. Sanguinetti MC, Jiang C, Curran mig, Keating MT. En mekanistisk länk mellan en ärftlig och en förvärvad hjärtarytmi: HERG kodar för IKr-kaliumkanalen. Cell (1995) 81:299-307. doi:10.1016/0092-8674(95)90340-2
Pubmed Abstrakt / Pubmed fulltext / CrossRef fulltext
8. Neyroud N, Tesson F, Denjoy I, Leibovici M, Donger C, Barhanin J, et al. En ny mutation i kaliumkanalgenen KVLQT1 orsakar Jervell och Lange-Nielsen kardioauditory syndrom. Nat Genet (1997) 15:186-9. doi: 10.1038 / ng0297-186
Pubmed Abstrakt / Pubmed fulltext / CrossRef fulltext
9. Kucera JP, Rohr S, Rudy Y. lokalisering av natriumkanaler i interkalerade skivor modulerar hjärtledning. Circ Res (2002) 91:1176-82. doi: 10.1161 / 01.RES.0000046237. 54156. 0 A
Pubmed Abstrakt / Pubmed fulltext / CrossRef fulltext
10. Oxford EM, Musa H, Maass K, Coombs W, Taffet SM, Delmar M. Connexin43 ombyggnad orsakad av hämning av plakofilin-2-uttryck i hjärtceller. Circ Res (2007) 101:703-11. doi: 10.1161/CIRCRESAHA.107.154252
Pubmed Abstrakt / Pubmed Fulltext / CrossRef Fulltext
11. Rohr S. Molekylär överhörning mellan mekaniska och elektriska korsningar vid den interkalerade skivan. Circ Res (2007) 101:637-9. doi: 10.1161/CIRCRESAHA.107.161901
CrossRef Fullständig Text
12. Det är en av de mest populära och mest populära. Återkommande intrauterin fosterförlust på grund av nära frånvaro av HERG: klinisk och funktionell karaktärisering av en homozygot nonsens HERG Q1070X-mutation. Hjärtrytm (2008) 5:553-61. doi: 10.1016 / j. hrthm.2008.01.020
PubMed Abstrakt / Pubmed Fulltext / CrossRef Fulltext
13. Bhuiyan ZA, Momenah TS, Amin TS, Al-Khadra AS, Alder M, Wilde AAM, et al. En Intronisk mutation som leder till ofullständig hoppning av exon-2 i KCNQ1 räddar hörsel i Jervell och Lange-Nielsen syndrom. Prog Biophys Mol Biol (2008) 98:319-27. doi: 10.1016/j.pbiomolbio.2008.10.004
PubMed Abstrakt / Pubmed Fulltext / CrossRef Fulltext
14. Bhuiyan ZA, Al-Shahrani S, Al-Khadra AS, Al-Ghamdi S, al-Kalaf K, Mannens MMAM, et al. Klinisk och genetisk analys av långt QT-syndrom hos barn från sex familjer i Saudiarabien: är de olika? Pediatric Cardiol (2009) 30:490-501. doi: 10.1007 / s00246-008-9377-y
PubMed Abstrakt / Pubmed fulltext / CrossRef fulltext
15. Al-Hazzani A, Dzimiri N, Tulbah S, Mallawi Y, al-Fayyadh M, et al. Identifiering av en ny kcnq1-mutation i en stor Saudisk Familj med långt QT-syndrom: kliniska konsekvenser och förebyggande konsekvenser. Clin Genet (2013) 83: 370-4. doi: 10.1111 / j. 1399-0004. 2012. 01914.X
PubMed Abstrakt / Pubmed fulltext / CrossRef fulltext
16. Schwartz PJ, Stramba-Badiale M, Crotti L, Pedrazzini M, Besana A, Bosi G, et al. Förekomst av det medfödda långa QT-syndromet. Cirkulation (2009) 120:1761-7. doi: 10.1161 / CIRCULATIONAHA.109.863209
PubMed Abstrakt / Pubmed Fulltext / CrossRef Fulltext
17. Schwartz PJ, Moss AJ, Vincent GM, Crampton RS. Diagnostiska kriterier för det långa QT-syndromet. Uppdatering. Cirkulation (1993) 88:782-4. doi: 10.1161 / 01.CIR.88.2.782
CrossRef Fullständig Text
18. Vi är ett av de mest populära företagen i världen. Sammanfattning: hrs/EHRA / APHRS konsensus uttalande om diagnos och hantering av patienter med ärftliga primära arytmisyndrom. Europace (2013) 15:1389-406. doi: 10.1093/europace / eut272
CrossRef Fullständig Text
19. Liu JF, Jons C, Moss AJ, McNitt S, Peterson DR, Qi M, et al. Syndrom Registret. Riskfaktorer för återkommande synkope och efterföljande dödliga eller nästan dödliga händelser hos barn och ungdomar med långt QT-syndrom. J Am Coll Cardiol (2011) 57:941-50. doi: 10.1016/j.jacc.2010.10.025
PubMed Abstrakt / Pubmed Fulltext / CrossRef Fulltext
20. Han är en av de mest kända i världen. Spektrum av mutationer i gener med långt QT-syndrom. KVLQT1, HERG, SCN5A, KCNE1 och KCNE2. Cirkulation (2000) 102:1178-85. doi: 10.1161 / 01.CIR.102.10.1178
Pubmed Abstrakt / Pubmed Fulltext / CrossRef Fulltext
21. Westenskow P, Splawski I, Timothy KW, Keating MT, Sanguinetti MC. Sammansatta mutationer: en vanlig orsak till allvarligt långt QT-syndrom. Cirkulation (2004) 109:1834-41. doi: 10.1161 / 01.CIR.0000125524.34234.13
Pubmed Abstrakt / Pubmed Fulltext / CrossRef Fulltext
22. Han är en av de mest kända och mest kända i världen. Risk för livshotande hjärthändelser hos patienter med långt QT-syndrom och multipla mutationer. Hjärtrytm (2013) 10:378-82. doi: 10.1016 / j. hrthm.2012.11.006
PubMed Abstrakt / Pubmed Fulltext / CrossRef Fulltext
23. Det är en av de viktigaste frågorna för dig. Genetisk testning i det långa QT-syndromet: utveckling och validering av ett effektivt tillvägagångssätt för genotypning i klinisk praxis. JAMA (2005) 294:2975-80. doi: 10.1001/jama.294.23.2975
PubMed Abstrakt / Pubmed Fulltext / CrossRef Fulltext
24. Roden DM. Klinisk praxis. Långt QT-syndrom. N Engl J Med (2008) 358:169-76. doi: 10.1056 / NEJMcp0706513
CrossRef Fullständig Text
25. Det är en av de viktigaste frågorna för dig. Genotyp-fenotypkorrelation i long-QT-syndromet: genspecifika triggers för livshotande arytmier. Cirkulation (2001) 103:89-95. doi: 10.1161 / 01.CIR.103.1.89
CrossRef Fullständig Text
26. Ackerman MJ, testare DJ, Porter CJ. Simning, en genspecifik arytmogen utlösare för ärftligt långt QT-syndrom. Mayo Clin Proc (1999) 74: 1088-94. doi:10.4065/74.11.1088
Pubmed Abstrakt / Pubmed fulltext / CrossRef fulltext
27. Det är en av de mest populära och mest populära spelen i världen. Genetisk testning för långt QT-syndrom: att skilja patogena mutationer från godartade varianter. Cirkulation (2009) 120:1752-60. doi: 10.1161 / CIRCULATIONAHA.109.863076
PubMed Abstrakt / Pubmed Fulltext / CrossRef Fulltext
28. Han är en av de mest kända och mest kända i världen. Kliniska aspekter av typ-1 Långt QT-syndrom efter plats, kodande typ och biofysisk funktion av mutationer som involverar kcnq1-genen. Cirkulation (2007) 115:2481-9. doi: 10.1161 / CIRCULATIONAHA.106.665406
Pubmed Abstrakt / Pubmed Fulltext / CrossRef Fulltext
29. Han är en av de mest kända och mest kända i världen. Varianter i den 3 ‘ oöversatta regionen av kcnq1-kodade Kv7.1 kaliumkanalen modifierar sjukdomens svårighetsgrad hos patienter med typ 1 Långt QT-syndrom på ett allelspecifikt sätt. Eur Hjärta J (2012) 33:714-23. doi: 10.1093/eurheartj / ehr473
Pubmed Abstrakt / Pubmed fulltext / CrossRef fulltext
30. Barsheshet A, Goldenberg I, O-Uchi J, Moss AJ, Jons C, Shimizu W, et al. Mutationer i cytoplasmatiska slingor i kcnq1-kanalen och risken för livshotande händelser: implikationer för mutationsspecifikt svar på terapi med blockerare i typ 1 Långt QT-syndrom. Cirkulation (2012) 125:1988-96. doi: 10.1161 / CIRCULATIONAHA.111.048041
Pubmed Abstrakt / Pubmed Fulltext / CrossRef Fulltext
31. L., L., L., L., L., L., L., L., L., L., L., L., L., L., L., L., L., L., L., L., L., L. Jervell och Lange-Nielsens syndrom: naturhistoria, molekylär grund och kliniskt resultat. Cirkulation (2006) 113:783-90. doi: 10.1161 / CIRCULATIONAHA.105.592899
PubMed Abstrakt / Pubmed Fulltext / CrossRef Fulltext
32. Bhuiyan ZA, Wilde AA. IKs i hjärta och hörsel, örat kan göra med mindre än hjärtat. Circ Cardiovasc Genet (2013) 6:141-3. doi: 10.1161 / CIRCGENETICS.113.000143
CrossRef Fullständig Text
33. Wollnik B, Schroeder BC, Kubisch C, Esperer HD, Wieacker P, Jentsch TJ. Patofysiologiska mekanismer för dominerande och recessiva KVLQT1 K + kanalmutationer som finns i ärftliga hjärtarytmier. Hum Mol Genet (1997) 6:1943-9. doi: 10.1093 / hmg / 6.11.1943
CrossRef Fullständig Text
34. Han är en av de mest kända i världen. Auditiva stimuli som utlösare för arytmiska händelser skiljer HERG-relaterade (LQTS2) patienter från KVLQT1-relaterade patienter (LQTS1). J Am Coll Cardiol (1999) 33:327-32. doi: 10.1016 / S0735-1097(98)00578-6
CrossRef Full Text
35. Han är en av de mest kända i världen. Ökad risk för arytmiska händelser vid långt QT-syndrom med mutationer i porregionen hos den humana eter-a-go-go-relaterade genkaliumkanalen. Cirkulation (2002) 105:794-9. doi: 10.1161 / hc0702. 105124
Pubmed Abstrakt / Pubmed fulltext / CrossRef fulltext
36. Lupoglazoff JM, Denjoy I, skurk E, Fressart V, Simon F, Bozio A, et al. Långt QT-syndrom hos nyfödda: ledningsstörningar associerade med HERG-mutationer och sinus bradykardi med kcnq1-mutationer. J Am Coll Cardiol (2004) 43:826-30. doi: 10.1016/j.jacc.2003.09.049
PubMed Abstrakt / Pubmed Fulltext / CrossRef Fulltext
37. P, Bellocq C, Millat G, Piqueras E, Potet F, Schott JJ, et al. Torsades de pointes komplicerar atrioventrikulärt block: bevis för en genetisk predisposition. Hjärtrytm (2007) 4:170-4. doi: 10.1016 / j. hrthm.2006.10.004
PubMed Abstrakt / Pubmed Fulltext / CrossRef Fulltext
38. Det är en av de mest populära och mest populära. Homozygot för tidig trunkering av HERG-proteinet: den mänskliga HERG-knockouten. Cirkulation (1999) 100:1264-7. doi: 10.1161 / 01.CIR.100.12.1264
Pubmed Abstrakt / Pubmed Fulltext / CrossRef Fulltext
39. Piippo K, Laitinen P, Swan H, Toivonen L, Viitasalo M, Pasternack M, et al. Homozygositet för en HERG kaliumkanalmutation orsakar en allvarlig form av långt QT-syndrom: identifiering av en uppenbar grundmutation hos finländarna. J Am Coll Cardiol (2000) 35:1919-25. doi: 10.1016 / S0735-1097(00)00636-7
Pubmed Abstrakt / Pubmed fulltext / CrossRef fulltext
40. Han är en av de mest populära och mest populära. Klinisk, genetisk och biofysisk karakterisering av en homozygot HERG-mutation som orsakar allvarligt neonatalt långt QT-syndrom. Pediatr Res (2003) 53:744-8. doi: 10.1203 / 01.PDR.0000059750.17002.B6
PubMed Abstrakt / Pubmed Fulltext / CrossRef Fulltext
41. Ackerman MJ, testare DJ, Jones GS, kommer ML, Burrow CR, Curran mig. Etniska skillnader i hjärtkaliumkanalvarianter: konsekvenser för genetisk mottaglighet för plötslig hjärtdöd och genetisk testning för medfödd långt QT-syndrom. Mayo Clin Proc (2003) 78:1479-87. doi:10.4065/78.12.1479
Pubmed Abstrakt / Pubmed fulltext / CrossRef fulltext
42. Crotti L, Lundquist AL, Insolia R, Pedrazzini M, Ferrandi C, De Ferrari GM, et al. KCNH2-K897T är en genetisk modifierare av latent medfödd långt QT-syndrom. Cirkulation (2005) 112:1251-8. doi: 10.1161 / CIRCULATIONAHA.105.549071
PubMed Abstrakt / Pubmed Fulltext / CrossRef Fulltext
43. Nof E, Cordeiro JM, p CJ, Scornik FS, Calloe K, Kärlek B, et al. En vanlig enkel nukleotidpolymorfism kan förvärra long-QT typ 2-syndrom som leder till plötslig spädbarnsdöd. Circ Cardiovasc Genet (2010) 3:199-206. doi: 10.1161 / CIRCGENETICS.109.898569
Pubmed Abstrakt / Pubmed Fulltext / CrossRef Fulltext
44. Bezzina C, Veldkamp MW, van Den Berg MP, Postma av, Rook MB, Viersma JW, et al. En enda na ( + ) kanalmutation som orsakar både lång-QT och Brugada syndrom. Circ Res (1999) 85:1206-13. doi: 10.1161 / 01.RES.85.12.1206
Pubmed Abstrakt / Pubmed fulltext / CrossRef fulltext
45. Kyndt F, Probst V, Potet F, Demolombe S, Chevallier JC, Baro I, et al. Ny SCN5A-mutation som leder antingen till isolerad hjärtledningsdefekt eller Brugadas syndrom i en stor fransk familj. Cirkulation (2001) 104:3081-6. doi: 10.1161 / hc5001. 100834
Pubmed Abstrakt / Pubmed fulltext / CrossRef fulltext
46. Det är en av de mest populära och mest populära. E1784k-mutationen i SCN5A är associerad med blandad klinisk fenotyp av typ 3 långt QT-syndrom. J Clin Investera (2008) 118:2219-29. doi: 10.1172 / JCI34057
Pubmed Abstrakt / Pubmed fulltext / CrossRef fulltext
47. Keller DI, Acharfi S, Delacr Exportaz E, Benammar N, Rotter M, Pfammatter JP, et al. En ny mutation i SCN5A, delQKP 1507-1509, vilket orsakar långt QT-syndrom: roll av q1507-rest i natriumkanalinaktivering. J Mol Cell Cardiol (2003) 35:1513-21. doi: 10.1016/j.yjmcc.2003.08.007
PubMed Abstrakt / Pubmed Fulltext / CrossRef Fulltext
48. Det finns många olika typer av produkter. Riskstratifiering i long-QT-syndromet. N Engl J Med (2003) 348:1866-74. doi: 10.1056 / NEJMoa022147
Pubmed Abstrakt / Pubmed fulltext / CrossRef fulltext
49. Lupoglazoff JM, Cheav T, Baroudi G, Berthet M, Denjoy I, Cauchemez B, et al. Homozygot SCN5A-mutation i långt QT-syndrom med funktionellt två-till-ett atrioventrikulärt block. Circ Res (2001) 89: E16-21. doi: 10.1161 / hh1401.095087
CrossRef Fullständig Text
50. Han är en av de mest kända och mest kända. Hjärtnatriumkanalmutationen delQKP 1507-1509 är associerad med det expanderande fenotypiska spektrumet av LQT3, ledningsstörning, utvidgad kardiomyopati och hög förekomst av plötslig död för Ungdomar. Europace (2008) 10:1329-35. doi: 10.1093 / europace / eun202
Pubmed Abstrakt / Pubmed fulltext / CrossRef fulltext
51. Det finns många olika typer av produkter. En heterozygot deletionsmutation i hjärtnatriumkanalgenen SCN5A med förlust – och förstärkningsfunktionsegenskaper manifesterar sig som isolerad ledningssjukdom, utan tecken på Brugada eller långt QT-syndrom. PLoS En (2013) 8:e67963. doi: 10.1371 / tidskrift.pone.0067963
PubMed Abstrakt / Pubmed Fulltext / CrossRef Fulltext
52. Växt LD, Bowers PN, Liu Q, Morgan T, Zhang T, Statliga MW, et al. En vanlig hjärtnatriumkanalvariant associerad med plötslig spädbarnsdöd hos afroamerikaner, SCN5A S1103Y. J Clin Invest (2006) 116:430-5. doi: 10.1172 / JCI25618
PubMed Abstrakt / Pubmed fulltext / CrossRef fulltext
53. Han är en av de mest kända och mest kända i världen. Ankyrin-B-mutation orsakar typ 4 lång-QT hjärtarytmi och plötslig hjärtdöd. Natur (2003) 421:634-9. doi: 10.1038 / nature01335
PubMed Abstrakt / Pubmed fulltext / CrossRef fulltext
54. Han är en av de mest kända i världen. Kartläggning av en gen för långt QT-syndrom till kromosom 4q25-27. Am J Hum Genet (1995) 57:1114-22.
PubMed Abstrakt / Pubmed Fulltext
55. Barhanin J, Lesage F, Guillemare E, Fink M, Lazdunski M, Romey G. K(V)LQT1 och LSK (minK) proteiner associerar för att bilda i(Ks) hjärtkaliumström. Natur (1996) 384:78-80. doi: 10.1038 / 384078a0
Pubmed Abstrakt / Pubmed fulltext / CrossRef fulltext
56. Det finns många olika typer av produkter. Coassembly av k (V)LQT1 och minK(IsK) proteiner för att bilda hjärt-i (Ks) kaliumkanal. Natur (1996) 384:80-3. doi: 10.1038 / 384080a0
PubMed Abstrakt / Pubmed fulltext / CrossRef fulltext
57. Schulze-Bahr E, Wang Q, Wedekind H, Haverkamp W, Chen Q, Sun Y, et al. Kcne1-mutationer orsakar Jervell och Lange-Nielsen syndrom. Nat Genet (1997) 17:267-8. doi: 10.1038 / ng1197-267
CrossRef Fullständig Text
58. Duggal P, Vesely MR, Wattanasirichaigoon D, Villafane J, Kaushik V, Beggs AH. Mutation av genen för IKs associerad med både Jervell och Lange-Nielsen och Romano-Ward former av långt QT-syndrom. Cirkulation (1998) 97:142-6. doi: 10.1186/1471-2350-9-24
Pubmed Abstrakt / Pubmed Fulltext / CrossRef Fulltext
59. Nishio Y, Makiyama T, Itoh H, Sakaguchi T, Ohno S, Gong YZ, et al. D85N, en kcne1-polymorfism, är en sjukdomsframkallande genvariant i långt QT-syndrom. J Am Coll Cardiol (2009) 54:812-9. doi: 10.1016/j.jacc.2009.06.005
PubMed Abstrakt / Pubmed Fulltext / CrossRef Fulltext
60. Han är en av de mest populära i världen. Genetiska variationer av kcnq1, KCNH2, SCN5A, KCNE1 och KCNE2 hos läkemedelsinducerade patienter med långt QT-syndrom. Mol Med (2004) 82: 182-8. doi: 10.1007 / s00109-003-0522-z
PubMed Abstrakt / Pubmed fulltext / CrossRef fulltext
61. Han är en av de mest kända i världen. MiRP1 bildar IKr kaliumkanaler med HERG och är associerad med hjärtarytmi. Cell (1999) 97:175-87. doi: 10.1016 / S0092-8674 (00)80728-X
Pubmed Abstrakt / Pubmed fulltext / CrossRef fulltext
62. Han är en av de mest kända och mest kända. En kcne2-mutation hos en patient med hjärtarytmi inducerad av auditiva stimuli och serumelektrolytobalans. Cardiovasc Res (2008) 77:98-106. doi: 10.1093 / CVR | cvm030
Pubmed Abstrakt / Pubmed fulltext / CrossRef fulltext
63. Gips NM, Tawil R, Tristani-Firouzi M, kan t, Bendahhou S, Tsunoda A, et al. Mutationer i Kir2.1 orsakar utvecklings-och episodiska elektriska fenotyper av Andersens syndrom. Cell (2001) 105:511-9. doi: 10.1016 / S0092-8674(01)00342-7
Pubmed Abstrakt / Pubmed Fulltext / CrossRef Fulltext
64. Kubo Y, Baldwin TJ, Jan YN, Jan LY. Primär struktur och funktionellt uttryck av en mus inåt likriktare kaliumkanal. Natur (1993) 362:127-33. doi: 10.1038 / 362127a0
Pubmed Abstrakt / Pubmed fulltext / CrossRef fulltext
65. Raab-Graham KF, Radeke CM, Vandenberg CA. Molekylär kloning och uttryck av ett mänskligt hjärta inåt likriktare kaliumkanal. Neuroreport (1994) 5:2501-5. doi: 10.1097/00001756-199412000-00024
Pubmed Abstrakt / Pubmed Fulltext / CrossRef Fulltext
66. Splawski I, Timothy KW, Sharpe LM, Decher N, Kumar P, Bloise R, et al. Ca (V)1.2 kalciumkanaldysfunktion orsakar en multisystemstörning inklusive arytmi och autism. Cell (2004) 119:19-31. doi: 10.1016/j.cell.2004.09.011
PubMed Abstrakt / Pubmed Fulltext / CrossRef Fulltext
67. Splawski I, Timothy KW, Decher N, Kumar P, Sachse FB, Beggs AH, et al. Allvarlig arytmistörning orsakad av hjärt-l-typ kalciumkanalmutationer. Proc Natl Acad Sci USA (2005) 102: 8089-96. doi: 10.1073/pnas.0502506102
PubMed Abstrakt / Pubmed Fulltext / CrossRef Fulltext
68. Gillis J, Burashnikov E, Antzelevitch C, Blaser S, Gross G, Turner L, et al. Lång QT, syndaktyly, gemensamma kontrakturer, stroke och ny CACNA1C-mutation: utvidgning av spektrumet av Timothy syndrom. Är J med Genet A (2012) 158A:182-7. doi: 10.1002 / ajmg.a.34355
PubMed Abstrakt / Pubmed Fulltext / CrossRef Fulltext
69. Dufendach KA, Giudicessi JR, Boczek NJ, Ackerman MJ. Maternal mosaicism förvirrar den neonatala diagnosen av typ 1 Timothy syndrom. Pediatrik (2013) 131:e1991–5. doi: 10.1542/peds.2012-2941
PubMed Abstrakt / Pubmed Fulltext / CrossRef Fulltext
70. Det är en av de mest populära och mest populära. Mutant caveolin – 3 inducerar ihållande sen natriumström och är associerad med långt QT-syndrom. Omsättning (2006) 114:2104–12. doi: 10.1161 / CIRCULATIONAHA.106.635268
Pubmed Abstrakt / Pubmed Fulltext / CrossRef Fulltext
71. Cronk LB, ni B, Kaku T, testare DJ, Vatta M, Makielski JC, et al. Ny mekanism för plötsligt spädbarnsdödssyndrom: ihållande sen natriumström sekundär till mutationer i caveolin-3. Hjärtrytm (2007) 4:161-6. doi: 10.1016 / j. hrthm.2006.11.030
PubMed Abstrakt / Pubmed Fulltext / CrossRef Fulltext
72. Han är en av de mest kända och mest kända i världen. Molekylär genetik hos caveolin – genfamiljen: konsekvenser för mänskliga cancerformer, diabetes, Alzheimers sjukdom och muskeldystrofi. Am J Hum Genet (1998) 63:1578-87. doi:10.1086/302172
CrossRef Full Text
73. Balijepalli RC, Foell JD, Hall DD, helvete JW, Kamp TJ. Lokalisering av hjärt-l-typ ca(2+) kanaler till ett caveolärt makromolekylärt signalkomplex krävs för beta (2) – adrenerg reglering. Proc Natl Acad Sci USA (2006) 103: 7500-5. doi: 10.1073/pnas.0503465103
PubMed Abstrakt / Pubmed Fulltext / CrossRef Fulltext
74. Det är en av de mest populära och mest populära spelen i världen. SCN4B-kodad natriumkanal beta4-subenhet vid medfödd långt QT-syndrom. Cirkulation (2007) 116:134-42. doi: 10.1161 / CIRCULATIONAHA.106.659086
Pubmed Abstrakt / Pubmed Fulltext / CrossRef Fulltext
75. Chen L, Marquardt ML, testare DJ, Sampson KJ, Ackerman MJ, Kass RS. Mutation av ett A-Kinas-förankringsprotein orsakar långt QT-syndrom. Proc Natl Acad Sci USA (2007) 104: 20990-5. doi: 10.1073/pnas.0710527105
PubMed Abstrakt / Pubmed Fulltext / CrossRef Fulltext
76. Wu G, Ai T, Kim JJ, Mohapatra B, Xi Y, Li Z, et al. Alfa-1-syntrofinmutation och det långa QT-syndromet: en sjukdom med natriumkanalstörning. Circ Arrythm Electrophysiol (2008) 1:193-201. doi: 10.1161/CIRCEP.108.769224
Pubmed Abstrakt / Pubmed Fulltext / CrossRef Fulltext
77. Yang Y, Yang Y, Liang B, Liu J, Li J, Grunnet M, et al. Identifiering av en Kir3.4 mutation vid medfödd långt QT-syndrom. Am J Hum Genet (2010) 86:872-80. doi: 10.1016/j.ajhg.2010.04.017
PubMed Abstrakt / Pubmed Fulltext / CrossRef Fulltext
78. Roden DM. Mekanismer och hantering av proarytmi. Am J Cardiol (1998) 82: 49I-57I. doi: 10.1016 / S0002-9149 (98)00472-X
CrossRef Fullständig Text
79. Mitcheson JS, Chen J, Lin M, Culberson C, Sanguinetti MC. En strukturell grund för läkemedelsinducerat långt QT-syndrom. Proc Natl Acad Sci USA (2000) 97:12329-33. doi: 10.1073/pnas.210244497
CrossRef Fullständig Text
80. Kannankeril PJ, Roden DM. Läkemedelsinducerad lång QT och torsade de pointes: senaste framstegen. Curr Opin Cardiol (2007) 22:39-43. doi: 10.1097 / HCO.0b013e32801129eb
PubMed Abstrakt / Pubmed fulltext / CrossRef fulltext
81. Det finns många olika typer av produkter. Långsam fördröjd likriktarkaliumströmblockad bidrar viktigare till läkemedelsinducerat långt QT-syndrom. Circ Arrythm Electrophysiol (2013) 6:1002-9. doi: 10.1161/CIRCEP.113.000239
PubMed Abstrakt / Pubmed Fulltext / CrossRef Fulltext
82. Sesti F, Abbott GW, Wei J, Murray KT, Saksena S, Schwartz PJ, et al. En vanlig polymorfism associerad med antibiotikainducerad hjärtarytmi. Proc Natl Acad Sci USA (2000) 97:10613-8. doi: 10.1073/pnas.180223197
PubMed Abstrakt / Pubmed Fulltext / CrossRef Fulltext
83. K. C. S., Crawford DC, Sinner MF, Behr er, Kannankeril PJ, Wilde AA, et al. En stor kandidatgenundersökning identifierar kcne1 d85n-polymorfismen som en möjlig modulator av läkemedelsinducerad torsades de pointes. Circ Cardiovasc Genet (2012) 5:91-9. doi: 10.1161 / CIRCGENETICS.111.960930
Pubmed Abstrakt / Pubmed Fulltext / CrossRef Fulltext
84. K, K, K, K, K, K, K, K, K, K, K, K, K, et al. Anti-kcnh2 antikroppsinducerat långt QT-syndrom: ny förvärvad form av långt QT-syndrom. J Am Coll Cardiol (2007) 50:1808-9. doi: 10.1016/j.jacc.2007.07.037
CrossRef Fullständig Text
85. Han är en av de mest kända och mest kända i världen. EKG t-vågmönster i genetiskt distinkta former av det ärftliga långa QT-syndromet. Cirkulation (1995) 92:2929-34. doi: 10.1161 / 01.CIR.92.10.2929
Pubmed Abstrakt / Pubmed Fulltext / CrossRef Fulltext
86. L. L., Timothy KW, Vincent GM, Lehmann MH, Fox J, Giuli LC, et al. Spektrum av St-t-vågmönster och repolariseringsparametrar vid medfödd långt QT-syndrom: EKG-fynd identifierar genotyper. Cirkulation (2000) 102:2849-55. doi: 10.1161 / 01.CIR.102.23.2849
PubMed Abstrakt / Pubmed Fulltext / CrossRef Fulltext
87. Han är en av de mest kända och mest kända i världen. Genotypspecifik uppkomst av arytmier vid medfödd långt QT-syndrom: möjliga terapikonsekvenser. Cirkulation (2006) 114:2096-103. doi: 10.1161 / CIRCULATIONAHA.106.642694
Pubmed Abstrakt / Pubmed Fulltext / CrossRef Fulltext
88. Det är en av de mest populära och mest populära. Ålders-och könsrelaterade skillnader i kliniska manifestationer hos patienter med medfödd långt QT-syndrom: resultat från International LQTS Registry. Cirkulation (1998) 97:2237-44. doi: 10.1161 / 01.CIR.97.22.2237
Pubmed Abstrakt / Pubmed Fulltext / CrossRef Fulltext
89. Han är en av de mest kända och mest kända i världen. Inverkan av genotyp på den kliniska kursen av long-QT-syndromet. International Long-QT-syndrom registret forskargrupp. N Engl J Med (1998) 339:960-5. doi: 10.1056 / NEJM199810013391404
PubMed Abstrakt / Pubmed fulltext / CrossRef fulltext
90. Goldenberg I, Moss AJ, Bradley J, Polonsky S, Peterson DR, McNitt S, et al. Långt QT-syndrom efter 40 års ålder. Cirkulation (2008) 117:2192-201. doi: 10.1161 / CIRCULATIONAHA.107.729368
Pubmed Abstrakt / Pubmed Fulltext / CrossRef Fulltext
91. Han är en av de mest populära och mest populära. Syndrom Registret. Modulerande effekter av ålder och kön på det kliniska förloppet av långt QT-syndrom efter genotyp. J Am Coll Cardiol (2003) 42: 103-9. doi: 10.1016 / S0735-1097(03)00554-0
Pubmed Abstrakt / Pubmed fulltext / CrossRef fulltext
92. Han är en av de mest kända och mest kända i världen. Långt QT-syndrom och graviditet. J Am Coll Cardiol (2007) 49:1092-8. doi: 10.1016/j.jacc.2006.09.054
PubMed Abstrakt / Pubmed Fulltext / CrossRef Fulltext
93. Schwartz PJ, Priori SG, Dumaine R, Napolitano C, Antzelevitch C, Stramba-Badiale M, et al. En molekylär länk mellan det plötsliga spädbarnsdödssyndromet och det långa QT-syndromet. N Engl J Med (2000) 343:262-7. doi: 10.1056 / NEJM200007273430405
CrossRef Fullständig Text
94. Det är en av de mest populära och mest populära. Molekylär diagnos hos ett barn med plötsligt spädbarnsdödssyndrom. Lancet (2001) 358:1342-3. doi: 10.1016 / S0140-6736(01)06450-9
Pubmed Abstrakt / Pubmed fulltext / CrossRef fulltext
95. L. A., L., L., L., L., L., L., L., L., L., L., L., L., L., L., L., L., L., L., L., L., L. Mutationer i HERG K + – jonkanalen: en ny länk mellan Långt QT-syndrom och plötsligt spädbarnsdödssyndrom. Am J Cardiol (2005) 95:433-4. doi: 10.1016 / j.amjcard.2004.09.054
PubMed Abstrakt / Pubmed Fulltext / CrossRef Fulltext
96. Testare DJ, Ackerman MJ. Postmortem långt QT-syndrom genetisk testning för plötslig oförklarlig död hos unga. J Am Coll Cardiol (2007) 49: 240-6. doi: 10.1016/j.jacc.2006.10.010
PubMed Abstrakt / Pubmed Fulltext / CrossRef Fulltext
97. Hofman N, Tan HL, Clur SA, Alders M, van Langen IM, Wilde AA. Bidrag av ärftlig hjärtsjukdom till plötslig hjärtdöd i barndomen. Pediatrik (2007) 120:e967–73. doi: 10.1542/peds.2006-3751
PubMed Abstrakt / Pubmed Fulltext / CrossRef Fulltext
98. Wedekind H, Smits JP, Schulze-Bahr E, Arnold R, Veldkamp MW, Bajanowski T, et al. De novo-mutation i SCN5A-genen associerad med tidig början av plötslig spädbarnsdöd. Cirkulation (2001) 104:1158-64. doi: 10.1161 / hc3501. 095361
CrossRef Fullständig Text
99. Wang DW, Desai RR, Crotti L, Arnestad M, Insolia R, Pedrazzini M, et al. Hjärtnatriumkanaldysfunktion vid plötsligt spädbarnsdödssyndrom. Cirkulation (2007) 115:368-76. doi: 10.1161 / CIRCULATIONAHA.106.646513
Pubmed Abstrakt / Pubmed Fulltext / CrossRef Fulltext
100. Det är en av de mest populära spelbolagen i världen. Molekylär och funktionell karakterisering av nya glycerol-3-fosfatdehydrogenas 1-liknande gen (GPD1-L) mutationer i plötsligt spädbarnsdödssyndrom. Cirkulation (2007) 116:2253-9. doi: 10.1161 / CIRCULATIONAHA.107.704627
Pubmed Abstrakt / Pubmed Fulltext / CrossRef Fulltext
101. Det är en av de mest populära och mest populära spelen i världen. Plötsliga spädbarnsdödssyndromassocierade mutationer i natriumkanalens beta-subenheter. Hjärtrytm (2010) 7:771-8. doi: 10.1016 / j. hrthm.2010.01.032
PubMed Abstrakt / Pubmed Fulltext / CrossRef Fulltext
102. Han är en av de mest kända och mest kända i världen. Återkommande fosterförlust i tredje trimestern och modermosaicism för långt QT-syndrom. Cirkulation (2004) 109:3029-34. doi: 10.1161 / 01.CIR.0000130666.81539.9 E
Pubmed Abstrakt / Pubmed Fulltext / CrossRef Fulltext
103. Han är en av de mest kända och mest kända i världen. Inte alla betablockerare är lika i hanteringen av långt QT-syndrom typ 1 och 2: högre återfall av händelser under Metoprolol. J Am Coll Cardiol (2012) 60:2092-6. doi: 10.1016/j.jacc.2012.07.046
PubMed Abstrakt / Pubmed Fulltext / CrossRef Fulltext
104. Schwartz PJ, Priori SG, Cerrone M, Spazzolini C, Odero A, Napolitano C, et al. Vänster hjärt sympatisk denervering vid hantering av högriskpatienter som drabbats av long-QT-syndromet. Cirkulation (2004) 109:1826-33. doi: 10.1161 / 01.CIR.0000125523.14403.1 E
Pubmed Abstrakt / Pubmed Fulltext / CrossRef Fulltext
105. Singh B, al Shahwan SA, Habbab MA, al deeb SM, Biary N. idiopatisk långt QT-syndrom: ställa rätt fråga. Lancet (1993) 341:741. doi:10.1016/0140-6736(93)90501-7
CrossRef Full Text
106. Gorgels AP, Al Fadley F, Zaman L, KANTOCH MJ, Al Halees Z. Det långa QT-syndromet med nedsatt atrioventrikulär ledning: en malign variant hos spädbarn. J Cardiovasc Electrophysiol (1998) 9:1225-32. doi: 10.1111 / j. 1540-8167. 1998.tb00096.X
PubMed Abstrakt / Pubmed fulltext / CrossRef fulltext
107. Kantoch MJ, Qurashi MM, Bulbul ZR, Gorgels AP. En nyfödd med en komplex medfödd hjärtsjukdom, atrioventrikulärt block och torsade de pointes ventrikulär takykardi. Pacing Clin Electrophysiol (1998) 21:2664-7. doi: 10.1111 / j. 1540-8159. 1998.tb00043.X
CrossRef Fullständig Text
108. El-Hazmi MA, al-Swailem AR, Warsy AS, al-Swailem AM, Sulaimani R, al-Meshari AA. Blodsband bland den saudiarabiska befolkningen. J Med Genet (1995) 32:623-6. doi: 10.1136/jmg.32.8.623
CrossRef Fullständig Text
109. El Mouzan MI, al Salloum AA, Al Herbish AS, Qurachi MM, Al Omar AA. Konsanguinitet och stora genetiska störningar hos saudiska barn: en samhällsbaserad tvärsnittsstudie. Ann Saudi Med (2008) 28:169-73. doi:10.4103/0256-4947.51726
Pubmed Abstrakt / Pubmed fulltext / CrossRef fulltext
110. Det är en av de mest populära och mest populära spelen i världen. RYR2-kodad ryanodinreceptor / kalciumfrisättningskanal hos patienter som tidigare diagnostiserats med antingen katekolaminerg polymorf ventrikulär takykardi eller genotyp negativ, träningsinducerat långt QT-syndrom: en omfattande mutationsanalys med öppen läsram. J Am Coll Cardiol (2009) 54:2065-74. doi: 10.1016/j.jacc.2009.08.022
PubMed Abstrakt / Pubmed Fulltext / CrossRef Fulltext
111. Al-Aama JY, Al-Ghamdi S, Bdier AY, Wilde AA, Bhuiyan ZA. De novo-mutation i kcnq1-genen som orsakar Jervell och Lange-Nielsens syndrom. Clin Genet (2013) (På Engelska). doi: 10.1111/cge.12300
PubMed Abstrakt / Pubmed Fulltext / CrossRef Fulltext
112. Vi är ett av de mest populära företagen i världen. Variant av SCN5A natriumkanal inblandad i risk för hjärtarytmi. Vetenskap (2002) 297:1333-6. doi: 10.1126/vetenskap.1073569
CrossRef Fullständig Text
113. Wilde AA, Bezzina CR. Genetik av hjärtarytmier. Hjärta (2005) 91:1352-8.