Mukopolysackaridoser: tidiga diagnostiska tecken hos spädbarn och barn
Mukopolysackaridoser (MPS) är en grupp kliniskt heterogena sjukdomar orsakade av brister i de lysosomala enzymer som krävs för nedbrytning av glykosaminoglykaner (GAGs).
MPS kännetecknas av progressiva och systemiska kliniska manifestationer. Trots deras biokemiska och genetiska heterogenitet delar olika typer viktiga kliniska egenskaper i olika kombinationer, inklusive LED-och skelettdysplasi med styvhet (utom MPS IV där det finns slapphet) och smärta, grova ansiktsdrag, hornhinnans grumling, inguinal eller bukbråck, återkommande övre luftvägsinfektioner, hjärtklaffsjukdom, karpaltunnelsyndrom och varierande neurologiskt engagemang. Dessa funktioner uppträder vanligtvis under de första månaderna av livet för de svåra formerna och i tidig barndom för de mer försvagade formerna, men underskattas ofta och beaktas i allmänhet endast när det är tydligt uppenbart.
sällsyntheten hos dessa störningar och variationen i klinisk presentation leder ofta till en diagnostisk fördröjning; detta kan sträcka sig från månader, för de svåra formerna, till år, för de försvagade (se Rigoldi et al. i detta tillägg). På grund av den snabba utvecklingen och det akuta behovet av intervention i de svåra presentationerna kan till och med månader av försening i diagnosen ge katastrofala resultat för patientens framtida hälsa.
vårt mål är att i denna recension understryka de mycket tidiga tecknen på de svåra formerna som bör varna barnläkaren vid deras första utseende.
vilka tecken / symtom kan vi förvänta oss hos barn med svåra former av MPS?
symtom och tecken som tyder på olika former av parlamentsledamöter rapporteras i Tabell 1 beroende på ålder. Under de första 6 månaderna av livet kan inguinalbråck, onormalt frekventa luftvägsinfektioner, otit och organomegali ses hos MPS-patienter, särskilt i MPS i (Hurler syndrom) som har den mest tidiga allvarliga starten. Dessutom, från 6 till 12 månader, utvecklar dessa spädbarn ofta en gibbus (toraco-lumbar kyphos), hjärtmumling, navelbråck, mild hypotoni och tillväxtfördröjning . De kan också ha det typiska grova ansiktsutseendet (Fig. 1). MPS II, VI och VII kan presentera med en liknande tidig fenotyp. MPS IV-patienter har en tidig skelettfenotyp med utseende av höftdysplasi under de första månaderna av livet och duva bröstet (pectus carinatum) under de första 12-18 månaderna. MPS II spädbarn närvarande med en liknande engagemang men med senare debut. Under det andra året av livet utvecklar mps i Hurler syndrom spädbarn kognitiv fördröjning medan en MPS II-patient kan identifieras endast på grund av överväxt, frekventa luftvägsinfektioner och flera operationer ; de typiska ansiktsegenskaperna visas först senare. Vissa MPS II-patienter utvecklar också hyperaktivitet mellan 18 och 24 månader. Under det tredje året av livet är hyperaktivitet och kognitiv fördröjning lätt igenkännliga i MPS II och MPS III.
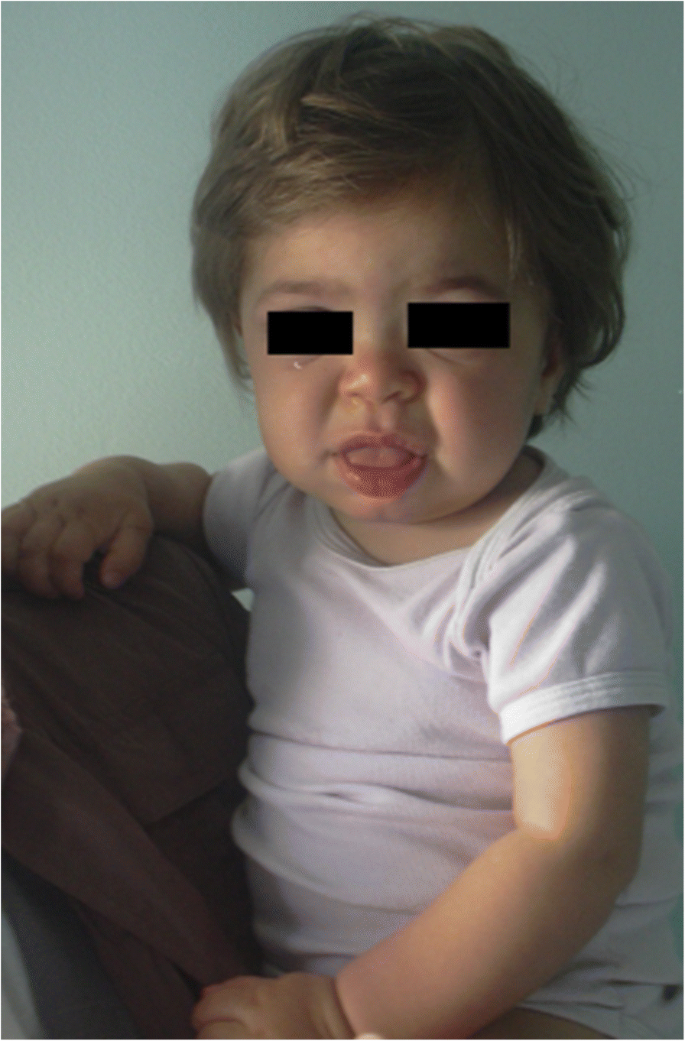
MPS IH i 1-årig patient. Grova facies: platt näsbrygga, makroglossi, frontal bossing
Sammanfattningsvis är skelettmanifestationer typiska och ibland äldre. Grova facies och kognitiv fördröjning är inte tidiga framträdande tecken.
vilka är de olika presentationerna av de olika typerna av parlamentsledamöter?
MPS med somatisk och kognitiv involvering
mukopolysackaridos typ i (MPS i; OMIM #252800) (Fig. 1, 2, 3, 4 och 5) orsakas av en brist på lysosomalt hydrolas, vilket leder till multisystemansamling av två GAGs: dermatansulfat (DS) och heparansulfat (HS) . Baserat på dess svårighetsgrad är MPS i traditionellt uppdelad i tre kliniska fenotyper; dessa former representerar emellertid ett kontinuum av sjukdomens svårighetsgrad. Hurlers syndrom, den allvarligaste formen, presenterar vanligtvis under det första leveåret. Berörda barn utvecklar snabbt signifikant kognitiv försämring och somatisk sjukdom i flera organsystem, vilket leder till döden inom det första decenniet i frånvaro av behandling. De försvagade formerna av MPS i, känd som Hurler–Scheie syndrom respektive Scheie syndrom, kännetecknas av senare symtom, längre livslängd och mild eller ingen inblandning i centrala nervsystemet (CNS). Arv är autosomalt recessivt, och i minst 50% av fallen är fenotypprediktion möjlig på basis av genotyp medan i de återstående 50% detekteras nya mutationer . Prevalensen är cirka 1 av 100 000 levande födda .

MPS IH. en Gibbus hos en 9 månader gammal patient. b ryggrad röntgen av ryggraden i en 3-årig patient. c T2-viktad magnetisk resonansbild av ryggraden som visar signifikant kyphos och ryggradsstenos

MPS IH. Röntgenbevis för dysostos multiplex. en förtjockning av revbenen i en 3-årig patient och B höftdysplasi hos en annan patient i åldern 18 månader

MPS IH: en klo hand i en 18 månader gammal patient; B hand röntgen.

MPS IH. en förtjockning av skallens kortikala ben och onormal “J-formad” konformation av sella turcica. Dilatation av periventrikulära utrymmen i b FLAIR och c T-2 viktade magnetiska resonansbilder av en patient i åldern 17 månader
mukopolysackaridos typ II (MPS II; OMIM #309900) (Fig. 6), även känt som Hunters syndrom, är resultatet av brist på enzymet iduronat-2-sulfatas (I2S) med följd av GAG-ackumulering av DS och HS . Prevalensen är cirka 1 av 140 000-156 000 manliga levande födda. I synnerhet är detta den enda X-länkade MPS, och kvinnliga bärare är vanligtvis asymptomatiska; de kan emellertid exceptionellt påverkas på grund av abnormiteter i X-kromosomen, homozygositeten eller skev-X-inaktivering . Det fenotypiska uttrycket spänner också över ett brett spektrum av klinisk svårighetsgrad. I svåra fall samexisterar markant progressivt neurologiskt engagemang och somatisk sjukdom, och döden inträffar vanligtvis under det andra decenniet av livet. Andra patienter med minimal eller ingen neurologisk dysfunktion kan uppvisa ett allvarligt somatiskt engagemang medan det i motsatt ände av spektrumet finns patienter med endast minimala somatiska manifestationer och normal intelligens som överlever till vuxen ålder . Tidiga tecken och symtom delas av de svåra formerna av MPS I och II.

parlamentsledamöter II. En 3-årig patient med endast milda karakteristiska ansiktsdrag
sporadiska fall av hydrops fetalis beskrivs i MPS I, även om dessa barn i allmänhet verkar normala vid födseln. De första tecknen på MPS jag brukar visas under de allra första månaderna av livet, medan de i MPS II brukar inträffa lite senare . Kiely et al. , i en retrospektiv studie på 55 Hurler-patienter, observerade att andningssymtom, utfodringssvårigheter, inguinalbråck och otitis media hade dykt upp vid en medianålder 6 månader i 6 månader, och kyfos, hjärtabnormaliteter, förstorad huvudomkrets, hypotoni, organomegali, hornhinnans grumling, ledbegränsning och navelbråck uppträdde mellan 6 och 12 månader. Navelbråck är en av de tidigaste presentationsfunktionerna i MPS I och II, och inguinalbråck rapporteras hos cirka 60% av patienterna .
en annan vanlig initial ledtråd för MPS I och II kan vara återkommande infektioner i övre luftvägarna med ökade utsöndringar. Frekvent otit, kronisk återkommande rinit och ihållande näsutsläpp utan uppenbar infektion är inte specifika, men kan hjälpa till att stärka den diagnostiska misstanken om den är förknippad med mer specifika tecken. Lagring av GAG i oro-svalget leder till utvidgning av tonsiller och polyper, och bidrar till övre luftvägskomplikationer. En annan frekvent upptäckt är bullrig andning, särskilt på natten, associerad i de senare stadierna med obstruktiv sömnapnoea. Som en följd av frekventa infektioner i mellanörat och dysostos i mellanöratets hörselben är hörselnedsättning frekvent, med både ledande och sensorineurala underskott . Tracheobronchomalacia observeras vanligtvis och kan leda till akut luftvägsobstruktion eller kollaps, med detta som en av de främsta dödsorsakerna under de följande åren .
adenoidektomi, tonsillektomi och tympanostomi, tillsammans med bråckreparation, är de mer frekventa och tidiga kirurgiska ingreppen för MPS i-och II-patienter, som ofta utförs innan de känner till diagnosen (i mer än 50% av fallen) .
återkommande diarre är ganska vanligt i tidiga stadier av MPS I och kan också förekomma hos mps II-patienter med neurologiskt engagemang, medan det är ovanligt hos de lätt drabbade patienterna .
gibbus deformitet (dorso-lumbar kyphos) (Fig. 2) blir kliniskt uppenbart vid en medianålder av 8.6 månader till 1 år i MPS I , som representerar en ganska specifik och äldre markör för MPS i allmänhet. Kotkropparna verkar tillplattad och näbb, potentiellt leder till senare komplikationer såsom spinal nerv infångning, akut ryggmärgsskada, och atlanto-occipital instabilitet; således, särskild försiktighet måste vidtas för att förhindra förskjutning av atlanto-axiell leden under narkos och kirurgi. Efter 1 års ålder representerar ledkontrakt och genu valgum andra frekventa tecken på nedsatt led och progressiv skelettdysplasi, som lätt kan upptäckas tidigare än 2 år hos alla spädbarn som drabbats av MPS. Alla dessa skelettmanifestationer tillsammans, typiska för MPS, heter dysostos multiplex. Förtjockning av revbenen kan ses på röntgenbilder (Fig. 3a), de långa benen är korta med breda axlar, och knäna är benägna att valgus och varus deformiteter. Phalangeal dysostos och synovial förtjockning leder till en karakteristisk klohanddeformitet (Fig. 4), vilket är en annan typisk egenskap som kan ge upphov till misstanke om sjukdomen. Höftdysplasi är ofta orsaken till att dessa barn först kan ses av ortopediska specialister. Vanligtvis är bäckenet dåligt format, lårhuvudena är små och coxa valga är vanligt, med progressiv och försvagande höftdeformitet (Fig. 3b).
det finns en märkbar skillnad mellan MPS i och MPS II; i MPS i bromsar skeletttillväxten med 3 års ålder, medan MPS II-patienter visar överväxt under de första 5-6 åren , vilket saktar tillväxten därefter .
grovbildning av ansiktsegenskaperna (bred näsa med utsvängda näsborrar, framträdande supraorbitala åsar, stora rundade kinder, tjocka läppar, förstorad utskjutande tunga) orsakad av lagring av GAGs i mjuka vävnader i orofacialregionen och ansiktsbenen blir uppenbara inom de första 2 åren, vid en medianålder på 1 , 1 år för Hurler-sjukdom och mellan 18 månader och 4 år i den allvarliga formen av Hunter-sjukdom, även om subtila fenotypiska förändringar kan detekteras tidigare (så tidigt som andra halvåret av det första året) av barnläkare med specifik erfarenhet (fig. 1). Makrocefali blir gradvis tydligare och scaphocefaly är vanligt (Fig. 5), men mikrocefali är också möjligt hos Hurler-patienter . Ansikts-och kroppshirsutism är alltid uppenbara vid 24 års ålder . Huden kan vara förtjockad och oelastisk. I synnerhet utvecklar vissa MPS II-patienter en distinkt hudskada, som beskrivs som elfenben-vita papler (stenliknande) på övre rygg och sidor av överarmarna, patognomonisk av Hunters syndrom .
kardiomyopati och progressiv förtjockning och förstyvning av ventilbladen hos MPS i-patienter kan leda till mitral och, mindre ofta, aortauppblåsning . Ett murmur kan detekteras under de första månaderna av livet och har ibland rapporterats som det första tecknet på sjukdomen . Hjärtinvolvering är också närvarande hos nästan alla patienter med MPS II, men detta börjar vid cirka 5 års ålder . Ventilsjukdom kan leda till höger och vänster ventrikulär hypertrofi och hjärtsvikt.
utbuktning av buken orsakad av progressiv hepatosplenomegali är lätt uppenbar efter 6 månaders ålder, även om lagring av GAGs i levern och mjälten inte leder till organdysfunktion .
Corneal clouding förekommer hos nästan alla individer med MPS I tidigare än 15 månaders ålder , vilket kan orsaka allvarlig synskada. Tvärtom är hornhinnans grumling inte ett typiskt inslag i Hunters syndrom , men slitslampaundersökning kan avslöja diskreta hornhinneskador som inte påverkar synen. Retinal dysfunktion kan detekteras, medan glaukom inte är ett vanligt resultat.
tidig psykomotorisk utveckling upptäcks vanligtvis som normalt, men i MPS i är utvecklingsförsening vanligtvis uppenbar vid 18 månaders ålder, med en progressiv mental nedgång som leder till en allvarlig intellektuell funktionsnedsättning. Språkkunskaper är mycket begränsade hos dessa barn. En global fördröjning av utvecklingsmilstoler i MPS II blir tydlig från 2 år framåt . Den ledande neurologiska egenskapen hos svår Hunters sjukdom representeras emellertid av markerade beteendestörningar, såsom hyperaktivitet, envishet och aggressivitet, som vanligtvis inte observeras hos dem med den försvagade fenotypen .
mukopolysackaridos typ VII (MPS VII; OMIM #253220), även känt som Sly-syndrom, är en extremt sällsynt MPS som kännetecknas av bristande aktivitet av Bisexuell glukuronidas, med lysosomal lagring av kondroitinsulfat (CS), DS och HS, vilket leder till cellulär och organdysfunktion. Den första patienten beskrevs av Sly et al. 1973 och totalt 145 patienter har rapporterats i litteraturen hittills.
den kliniska presentationen och sjukdomsprogressionen av MPS VII spänner över ett brett allvarlighetsspektrum, från tidiga, allvarliga multisystemmanifestationer och död under de allra första månaderna, till en mildare fenotyp med senare debut, normal eller nästan normal intelligens och längre överlevnad .
Fenotypegenskaper hos patienter med MPS VII liknar MPS i och MPS II (kort statur, skelettdysplasi, makrocefali, gingivalhypertrofi, återkommande öroninfektioner, hepatosplenomegali, bråck, hjärtinvolvering, nedsatt lungfunktion och kognitiv försämring). En oväntat hög andel av beskrivna patienter (41%) har en historia av icke-immun neonatal hydrops fetalis (NIHF) . Trots sjukdomens prenatala början hos dessa patienter överlevde 13 av 23 spädbarn med en mild till mellanliggande kurs i sena tonåren. Således förutsätter närvaron av NIHF inte i sig den möjliga svårighetsgraden av den kliniska kursen om patienten överlever spädbarn. Ett annat tidigt tecken är makrocrania, medan hjärtventilinvolvering och upprepade infektioner i övre och nedre luftvägarna uppträder under de första 2 åren av livet. Måttlig mental retardation och progressiv hörselnedsättning med talsvårigheter är också uppenbara med tiden.
Sammanfattningsvis har de svåra formerna av MPS jag en mycket tidig multisystemstart (inom 6 månader av livet); MPS II är liknande men med en senare debut, och MPS VII har en prenatal debut med frekvent NIHF och tidigt allvarligt engagemang i övre och nedre luftvägarna.
MPS med mestadels neurologiskt och kognitivt engagemang
Mukopolysackaridoser typ III (MPS III, även känt som Sanfilippo syndrom; Fig. 7) har en utbredd neurologisk presentation. MPS III är en lysosomal lagringsstörning orsakad av brist på ett av de fyra enzymerna som är involverade i katabolismen av HS. Fyra olika subtyper är kända—MPS III typ A (OMIM #252900), typ B (OMIM #252920), typ C (OMIM #252930) och typ D (omim #252940)—var och en på grund av en annan enzymbrist: typ A, sulfamidas/SGSH-gen; typ B, alfa-N-acetylglukosaminidas/NAGLU-gen; typ C, alfa-glukosaminid N-acetyltransferas/HGSNATGEN; typ D, n-acetilglukosamina-6-solfato sulfatasi/GNS-gen). Alla fyra typerna (A till D) har autosomalt recessivt arv. MPS III är den vanligaste av MPS med en uppskattad prevalens mellan 0.3 och 4.1 fall för varje 100 000 nyfödda, beroende på subtyp och ras .

parlamentsledamöter IIIA. Samma patient vid en 2,5 och b 5 år gammal. Notera det olika ansiktsuttrycket, som visar progression av den neurologiska försämringen
patienter med Sanfilippo syndrom visar progressiv kognitiv försämring under andra och tredje levnadsåret. Till skillnad från majoriteten av parlamentsledamöterna är somatiska egenskaper relativt mindre uppenbara och CNS-involveringen är iögonfallande. Talutvecklingsfördröjning är vanligtvis det första tecknet som föräldrarna märker men de viktigaste problemen kan vara beteendeproblem (hyperaktivitet, oroligt och aggressivt beteende, sömnstörningar), följt av progressiv mental nedgång . Dessa symtom kan orsaka feldiagnos som beteendestörningar, ADHD (attention deficit hyperactivity disorder) och autismspektrumstörningar . Epilepsi kan också vara närvarande.
sömnstörningar, vars förekomst rapporteras upp till 80-90% , är en vanlig egenskap. De består av svårigheter att somna och frekvent nattlig uppvaknande, med fullständig reversering av dag–nattrytmen hos vissa patienter.
alla dessa manifestationer är mycket svåra att hantera och har en enorm psykologisk inverkan på livskvaliteten för hela familjen.
i allmänhet, även om fenotypen kan vara mycket lika i de fyra undertyperna, verkar den kliniska kursen i Sanfilippo B och C kännetecknas av en mindre allvarlig fenotyp .
icke-neurologiska symtom är vanligtvis mindre uttalade hos MPS III än hos de andra MPS. Återkommande öron -, näs-och halsinfektioner observeras vid yngre ålder. Återkommande otit, defekter i mellanörat och avvikelser i innerörat kan orsaka dövhet. Ett överskott av tjocka sekret och anatomiska förändringar kan ge obstruktion av luftvägarna. Under de första åren av livet rapporteras diarre ofta, medan förstoppning är vanligare hos äldre patienter.
fysisk undersökning kan visa grova ansiktsdrag, även om dessa är mindre uttalade. Patienter har makrocefali, breda ögonbryn med medial flaring och synophrys, håret är vanligtvis torrt och grovt och hypertrichos är vanligt. Hos yngre patienter observeras ofta mild hepatomegali och navel-och inguinalbråck. Sällsynta och ofta milda kontrakturer finns oftast i armbågarna. Tillväxten påverkas hos ungefär hälften av patienterna äldre än 12 år .
Sammanfattningsvis har MPS III eller Sanfilippos sjukdom ett framträdande neurologiskt engagemang med mycket milda somatiska tecken. Småbarn i åldern 2-3 år som utvecklar hyperaktivitet, ADHD och aggressivt beteende kan misstänkas ha MPS III.
MPS med vanligt somatiskt engagemang
MPS IV och MPS VI närvarande med endast somatiskt engagemang. Dessa är progressiva förhållanden som sparar intellektuell förmåga och påverkar främst skelettet (MPS IVA) eller alla organ/system i kroppen (MPS VI). Arv är autosomalt recessivt. Den hastighet med vilken symtomen förvärras varierar mellan drabbade individer.
mukopolysackaridos IV (MPS IV, även känd som Morquio syndrom; Fig. 8) presenterar som två genetiskt distinkta störningar, var och en med olika enzymbrist: MPS IVA (Morquio a-syndrom; omim #253000) på grund av brist på N-acetylgalaktosamin-6-sulfatas (GALNS-gen), vilket resulterar i ackumulering av keratansulfat (KS) och CS ; och MPS IVB (Morquio B-syndrom; OMIM # 253010) på grund av brist på beta-galaktosidasaktivitet som leder till urin KS och oligosackaridutskiljning som hos GM1-patienter. MPS IVB är verkligen allelisk för de olika formerna av GM1-Gangliosidos, men saknar den psykomotoriska försämringen som ses vid GM1 Gangliosidos .

riksdagsledamöter IVA. en 4-årig patient med b antero-posterior och C laterala röntgenstrålar som visar “paddelformade” revben och platta och rundade ryggkotor med en typisk” främre näbb ” – aspekt
patienter med svår MPS IVA utan detekterbar enzymaktivitet avslöjar sina första symtom vanligtvis under det första leveåret, även om de sällan diagnostiseras före 4-5 års ålder. Information om Morquio a-patienternas naturhistoria härrör huvudsakligen från två omfattande datasamlingar . Minskad tillväxttakt börjar vid ungefär 18 månaders ålder och tillväxten stannar vid ungefär 7 eller 8 års ålder. Till skillnad från de andra parlamentsledamöterna är Morquio a-lederna vanligtvis slappa och mycket flexibla (hypermobila), men vissa leder (vanligtvis höfter och axlar) kan ha ett begränsat rörelseområde. Patienter med sådant skelett involvering kan få en diagnos av, eller genomgå utvärdering för, Spondyloepifyseal dysplasi, pseudoachondroplasi, multipel epifyseal dysplasi, eller bilateral Legg-Calv Baccarat-Perthes sjukdom .
en konstant funktion är odontoid hypoplasi; således kan dislokation av den atlanto-occipitala leden uppstå och orsaka kompression och skada på bulbar och ryggmärg, vilket resulterar i förlamning eller till och med död om cervikal stabilisering inte utförs tidigt.
MPS IVB-patienter har en mycket liknande klinisk manifestation men med en mindre framträdande kort statur. De har milt grova ansiktsdrag, hörselnedsättning, hornhinnans opacitet, valvulär hjärtsjukdom och frekventa övre luftvägsinfektioner. De kan också presentera med inguinal bråck och mild hepatomegali. Skelettdysplastiska funktioner inkluderar platyspondyly, odontoid hypoplasi med möjlig cervikal subluxation, kypho-skolios, coxa valga och trånga iliacvingar .
mukopolysackaridos VI (MPS VI, även känd som Maroteaux-Lamy syndrom; OMIM #253200; Fig. 9) beror på patogena mutationer i arylsulfatas B (ARSB) – genen belägen på kromosom 5. Som en konsekvens, minskad eller frånvarande aktivitet av enzymet arylsulfatas B (ASB) (N-acetylgalaktosamin 4-sulfatas) försämrar nedbrytning av DS bestämma dess cellulära ackumulering .

MPS VI. en 2-årig pojke med uppenbar grov ansikte och skelettdysostos
patienter utan detekterbar enzymaktivitet har den allvarliga formen av MPS VI och uppvisar symtom i tidig barndom. Deras somatiska fenotyp liknar Hurler syndrom. I de flesta fall, vid 2 eller 3 års ålder, blir allvarlig och progressiv skelettskada, kallad dysostos multiplex, uppenbar. Denna skelettdysplasi inkluderar höftdysplasi med dysplastiskt lårbenshuvud, onormala ryggkroppar, oregelbundna nyckelben, hypoplastisk distal ulna och radie och dysplastiska och korta metakarpala ben; karpala och tarsala ben är hypoplastiska och har en oregelbunden profil. Tillväxthastigheten saktar ofta efter det första levnadsåret, med en slutlig höjd i allmänhet lägre än 120 cm. Som i andra parlamentsledamöter är ett tidigt tecken grova ansiktsdrag. Dessutom har patienter en typisk utskjutande buk, hepatomegali, navel-och/eller inguinalbråck och hjärtinvolvering i tidig spädbarn. Även om ingen primär intellektuell funktionsnedsättning finns, kan hydrocephalus ses hos vissa patienter med därmed intrakraniell hypertoni och papilledema. Pectus carinatum, styva och kontraherade leder, skolios och kyphos och cervikal ryggmärgskompression förvärras med tiden .
Sammanfattningsvis visar MPS IV och VI inte CNS-involvering. MPS IV är en unik MPS för att visa gemensam slapphet, medan svår MPS VI-debut liknar den som observerats i MPS I.