Hranicích v Genetice
Úvod
Buněčné procesy jsou poháněny více interagujících molekul, jejichž úroveň aktivity musí být dynamicky regulována (Kitano, 2002). Výsledkem bude, že geny patřící do stejné signální a metabolické dráhy nebo sdílení podobných funkcí budou mít tendenci být koexprimovány napříč podmínkami (Wang et al., 2016). Ko-exprese genu modul analýzy vytváří sítě tvořené sady genů (tj. moduly), jejichž exprese je vysoce korelované. Tato analýza byla použita k odhalení funkčních modulů souvisejících s infekčními (Janova et al ., 2015), zánětlivé (Beins et al., 2016) a neurologické (Voineagu et al., 2011) nemoci, stejně jako několik typů rakoviny (Sharma et al., 2017).
Vážený gen co-výraz síťové analýzy (WGCNA) je široce používaná metoda k identifikaci co-vyjádřil genové moduly (Zhang a Horvath, 2005). Aby bylo možné spustit WGCNA, nicméně uživatelé jsou povinni být obeznámeni s programovací prostředí, stejně jako ručně vybrat parametry. Tyto vlastnosti brání vědcům s nedostatečnou znalostí R identifikovat genové moduly z transkriptomových datových sad.
na základě našeho Bioconductor R balíčku s názvem CEMiTool (Russo et al ., 2018), vyvinuli jsme uživatelsky přívětivou webovou aplikaci, která umožňuje vědcům bez zázemí v bioinformatice provádět komplexní analýzu koexpresní sítě.
Materiály a Metody
webové rozhraní webCEMiTool byl vyvinut s cílem umožnit uživatelům rychle vytvářet komplexní analýzy bez nutnosti instalace jakéhokoli specifického programu nebo internetového prohlížeče. Jediným požadavkem pro spuštění modulární analýzy je soubor dat obsahující úrovně exprese všech genů ve vzorcích za různých biologických podmínek (zde definovaných jako “třídy”). Neexistuje žádný definovaný rozsah počet vzorků, ale naše předchozí studie naznačuje minimálně 15 vzorků na sadu dat (Russo et al ., 2018). RNA-seq nebo microarrays), může být také potenciálně použit pro identifikaci modulů proteinů, cytokinů a dokonce i metabolitů. webCEMiTool poté automaticky vybere vstupní geny a identifikuje moduly koexprese. Každý modul obsahuje sadu genů, jejichž exprese sleduje podobný vzorec.
v rámci webCEMiTool jsme implementovali funkci, která hodnotí aktivitu genových modulů na každé třídě vzorků. Za tímto účelem musí uživatelé poskytnout pouze textový soubor oddělený tabulátorem anotace vzorku, který informuje třídu každého vzorku. “Profilový graf” zobrazující střední úroveň jednotlivých genů v modulu je pak zobrazen v části” výsledky ” nástroje (obrázek 1A).
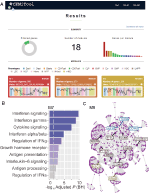
Obrázek 1. webCEMiTool přehled. (A) souhrn výsledků webCEMiTool-graf koblih představuje podíl vybraných genů bez dozoru filtrem. Přední strana také zobrazuje počet získaných modulů a sloupcový graf zobrazující počet genů v každém modulu. Grafy profilů modulů ilustrují střední expresi genů z modulů napříč každým vzorkem. Barvy představují různé třídy vzorků. (B) nepoměrně vysoké analýzu – To líčí −log10 upravená p-hodnota (Benjamini-Hochberg) obohacený drah v modulu (cesty definované uživatelem-vloženy .soubor gmt). (C) Gen sítě modul – top nejvíce spojeny geny (uzly) jsou označeny a barevné podle toho, zda byly původně přítomny v modulu (modrá), nebo je vložen z uživatelem zadaných interakce soubor (červený), nebo obojí (zelená).
umožnit funkční analýzy, uživatelé mohou také zkontrolovat, zda gen moduly jsou spojeny s konkrétní signalizační nebo metabolických drah (Obrázek 1B). Tyto cesty lze snadno extrahovat z databází, jako jsou KEGG, Reactome a MySigDB. Konečně, mohou uživatelé integrovat výsledky s interactome data (např. protein-protein interakcí, transkripční faktory a jejich transkribovaných genů, nebo dokonce miRNAs a jejich cílové geny). Tato funkce umožňuje uživatelům identifikovat kritické regulátory modulů (Obrázek 1C), poskytuje cenné poznatky pro experimentální ověření nebo potenciální cíle pro léky. Další podrobnosti o tom, jak získat volitelné soubory, naleznete na stránce “tutoriál” na webu.2
prokázat, že naše metoda je robustní, provedli jsme bezprecedentní ve velkém měřítku modulární analýzy s více než 1000 veřejně dostupných RNA-seq a microarray data sady a nové RNA-seq dat pacientů infikovaných Leishmania pomocí CEMiTool R package version (Russo et al., 2018). Přestože webCEMiTool a balíček mají odlišné vizualizační funkce a jsou založeny na různých platformách, základní funkce co-expression je v podstatě stejná. On-line nástroj popisujeme zde je postavena tak, aby umožňují snadný přístup k gene modulární analýzy pro non-programování vědci, zatímco R library verze je zaměřena na uživatele s větší znalostí z R programovací jazyk. Panel výsledků se navíc skládá z interaktivních grafů, které usnadňují interpretaci. Navíc s využitím rostoucího ekosystému webových služeb bioinformatiky vytváří náš nástroj rozhraní s platformou Enrichr (Chen et al ., 2013), umožňující bohatší zážitek pro naše uživatele.
výsledky
ukázali jsme, že webCEMiTool lze použít k analýze expresních dat na úrovni jedné buňky. Veřejně dostupné viscRNA-Seq dat (virus-včetně jedné buňky RNA-Seq) byly získány z NCBI GEO databáze (přístupové číslo GSE110496) a použita jako vstup pro analýzu. Údaje se vztahují k transcriptome jednotlivých lidského hepatomu (Huh7) buněk, které byly infikovány buď dengue virus (DEN) nebo Zika virus (ZIKV), pomocí multiplicita infekce (MOI) 0, 1, nebo 10 (Zanini et al., 2018). Buňky získané na čtyřech různých časových bodech (4, 12, 24 a 48 h po infekci) pak byly řazeny na jedné buňky transcriptomic analýza s upravenou Smart-seq2 protokolu (Zanini et al., 2018). V DEN data set se skládá 933 infikovaných buněk (MOI = 1 nebo 10) a 303 kontrol (MOI = 0), zatímco ZIKV data set se skládá z 488 infikovaných buněk (MOI = 1) a 403 kontrol. Před předložením analýzy webCEMiTool platformy, obě sady dat byly log10 transformovaných a geny, které nebyly vyjádřeny ve více než 80% vzorků byly odstraněny. Datové sady byly poté rozděleny podle viru a časového bodu a použity jako vstup (pole” Expression file”) do webCEMiTool. Kromě údajů o genové expresi jsme webCEMiTool poskytli také fenotypy vzorků (tj., virové zátěže) a Reaktomové genové sady.
naše analýzy webCEMiTool generovaly v průměru šest modulů za časový bod infekce DENV a více než osm modulů za časový bod infekce ZIKV. Vybrali jsme jeden modul za časový bod jako zástupce našich zjištění (obrázek 2A). Je zřejmé, že po 24 a 48 hodinách po infekci se expresivní aktivita reprezentativních modulů zvyšuje podle virové zátěže (obrázek 2A). Dále jsme provedli analýzu obohacení cesty reprezentativních modulů v 24 h po infekci pomocí odkazu webCEMiTool pro Enrichr (obrázek 2B). Tato zjištění nejen potvrzují to, co bylo popsáno v původní publikaci (Zanini et al ., 2018), ale také poskytnout nové poznatky o fyziopatologii infekcí virem dengue a Zika.
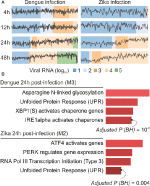
Obrázek 2. webCEMiTool aplikován na jednobuněčné RNA-seq dat. A) profilový graf společně exprimovaných genových modulů. Vybrali jsme jeden reprezentativní modul pro každý časový bod po infekci virem dengue (vlevo) nebo po infekci virem Zika (vpravo). Černá čára představuje střední expresi aktivity genů z modulů napříč každým vzorkem. Barvy představují různé množství virové RNA v buňce. (B) analýza nadměrné reprezentace vybraných modulů po 24 hodinách po virové infekci. Sloupcové grafy byly upraveny z Enrichr webtool spojené s webCEMiTool. Tyče jsou úměrné −log10 upravené p-hodnoty (Benjamini-Hochberg) obohacených drah v modulu.
Diskuse
i když několik podobných webových aplikací byly vyvinuty k provádění co-vyjádření genové analýzy (Tzfadia et al., 2016; Desai a kol., 2017), tyto nástroje neposkytují srovnatelné výsledky s webCEMiTool. Jednou z takových aplikací je GeNET (Desai et al., 2017). Tento webtool byl navržen tak, aby usnadnil genovou koexpresi analýzy a poskytuje analýzu obohacení a Gen-to-genové sítě. Tyto analýzy však provádí pouze u tří organismů (R. capsulatus, m. tuberculosis a o. sativa). Dalším příkladem je CoExpNetViz (Tzfadia et al., 2016), webový nástroj určený pro vizualizaci a konstrukci genových sítí. Podobně jako GeNET, CoExpNetViz je poněkud omezený s ohledem na organismy, protože se uvádí, že je primárně určen pro rostlinné transkriptomy. WebCEMiTool si klade za cíl poskytnout koexpresní analýzy pro jakýkoli organismus. Přestože je CoExpNetViz prezentován jako webová aplikace, jeho výsledky jsou uživatelům vráceny jako komprimovaná složka obsahující README.txt soubor s pokyny, jak vizualizovat své výsledky na Cytoscape app. Uživatelé pak musí ručně vložit do Cytoscape několik různých výstupních souborů poskytovaných nástrojem. Tyto další kroky mohou také způsobit, že proces je náchylný k chybám a možná skličující pro uživatele, kteří nejsou obeznámeni s Cytoscape. WebCEMiTool nabízí mnohem pohodlnější výsledky zobrazené v prohlížeči.
také jsme ukázali, že webCEMitool je schopen analyzovat jednobuněčná RNA-seq data rychleji a efektivněji. Naše výsledky vrátily relevantní informace o biologických procesech spojených s infekcí virem dengue a Zika. Všechny tyto analýzy byly provedeny v automatizované a praktickým způsobem, bez nutnosti uživatel mít hluboké pochopení pro vnitřní zpracování genové co-analýzu dat exprese.
Autor Příspěvků
LC, PR, BG-C, a MA-P provedené analýzy. LC, GS-H a VM-C vyvinuly webtool. HN tento nástroj vymyslely a na práci dohlížely. Všichni autoři pomáhají při psaní příspěvku.
Střet Zájmů Prohlášení
autoři prohlašují, že výzkum byl prováděn v nepřítomnosti jakékoli obchodní nebo finanční vztahy, které by mohlo být chápáno jako potenciální konflikt zájmů.