Fronteiras em Genética
Introdução
processos Celulares são conduzidos por várias moléculas interagindo cujo nível de atividade deve ser dinamicamente regulamentado (Kitano, 2002). Como resultado, genes pertencentes à mesma via de sinalização e metabólica ou compartilhando funções semelhantes tendem a ser co-expressos através de condições (Wang et al., 2016). A análise do módulo genético de co-expressão cria redes que compreendem conjuntos de genes (ou seja, módulos) cuja expressão está altamente correlacionada. Esta análise foi aplicada para revelar módulos funcionais relacionados com infecciosos (Janova et al., 2015), inflammatory (Beins et al., 2016), e neurológico (Voineagu et al., 2011) doenças, bem como vários tipos de câncer (Sharma et al., 2017).
Wgcna (Weighted gene co-expression network analysis) é um método amplamente utilizado para identificar módulos de genes Co-expressos (Zhang e Horvath, 2005). A fim de executar WGCNA, no entanto, os usuários são obrigados a estar familiarizados com ambientes de programação, bem como para selecionar manualmente parâmetros. Estas características impedem os investigadores com conhecimentos insuficientes de R de identificar módulos genéticos a partir de conjuntos de dados transcriptomas.
Based on our Bioconductor R package named Ceitool (Russo et al., 2018), desenvolvemos uma aplicação de fácil utilização baseada na web que permite aos cientistas sem experiência em Bioinformática realizar uma análise abrangente da rede de co-expressão.
materiais e métodos
a interface web do webCEMiTool foi desenvolvida para permitir aos usuários gerar rapidamente análises abrangentes sem a necessidade de instalar qualquer programa específico ou navegador de internet. O único requisito para executar a análise modular é um conjunto de dados contendo os níveis de expressão de todos os genes em amostras sob diferentes condições biológicas (aqui definido como “classes”). Não existe um número definido de amostras, mas o nosso estudo anterior sugere um mínimo de 15 amostras por conjunto de dados (Russo et al., 2018). Embora tenha sido concebido principalmente para dados de transcriptoma (isto é, RNA-seq ou microarrays), também pode ser usado potencialmente para identificar módulos de proteínas, citocinas e até mesmo metabolitos. webCEMiTool irá então selecionar automaticamente os genes de entrada e identificar os módulos de co-expressão. Cada módulo contém um conjunto de genes cuja expressão segue um padrão similar.
implementámos, no webCEMiTool, uma funcionalidade que avalia a actividade dos módulos genéticos em cada classe de amostras. Para isso, os usuários só têm que fornecer um arquivo de texto delimitado por tabulação de amostra que informa a classe de cada amostra. Na secção” resultados “da ferramenta (figura 1A) é apresentado um” gráfico de perfil ” que mostra o nível médio dos genes individuais dentro do módulo.
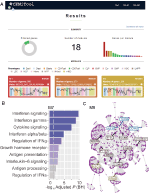
Figura 1. webcemitool overview. (A) resumo dos resultados webCEMiTool – o gráfico do donut representa a proporção de genes seleccionados pelo filtro não supervisionado. A primeira página também mostra o número de módulos obtidos, bem como um gráfico de barras representando o número de genes em cada módulo. As parcelas do perfil do módulo ilustram a actividade de expressão mediana dos genes a partir dos módulos em cada amostra. As cores representam as diferentes classes de amostra. B) análise da Sobrerepresentação, que apresenta o valor – p ajustado log10 (Benjamini −Hochberg) das vias enriquecidas num módulo (vias definidas por utilizadores inseridos .ficheiro gmt). (C) Gene network of a module – The top most connected genes (hubs) are labeled and colored based on whether they were originally present in the module (blue), or inserted from a user-inputted interaction file (red), or both (green).
para permitir a análise funcional, os usuários também podem verificar se os módulos do gene estão associados a vias de sinalização ou metabólicas específicas (figura 1B). Estas vias podem ser facilmente extraídas de bases de dados, como KEGG, Reactome e MySigDB. Finalmente, os usuários podem integrar os resultados com dados interactômicos (ou seja, interações proteína-proteína, fatores de transcrição e seus genes transcritos, ou mesmo miRNAs e seus genes alvo). Esta característica permite que os utilizadores identifiquem reguladores críticos dos módulos (figura 1C), fornecendo informações valiosas para a validação experimental ou alvos potenciais para os medicamentos. Detalhes adicionais sobre como obter os arquivos opcionais podem ser encontrados na página “Tutorial” do site.2
para demonstrar que o nosso método é robusto, realizámos uma análise modular em larga escala sem precedentes com mais de 1. 000 conjuntos de dados RNA-seq e microarray acessíveis ao público e novos dados RNA-seq de doentes infectados com Leishmania utilizando a versão da embalagem Ceitool R (Russo et al., 2018). Embora o webCEMiTool e o pacote tenham características de visualização distintas e sejam baseados em diferentes plataformas, a funcionalidade de co-expressão do núcleo é essencialmente a mesma. A ferramenta on-line que estamos descrevendo aqui é construída para permitir um fácil acesso a análises modulares de genes para pesquisadores não-programadores, enquanto a versão R Biblioteca é voltada para usuários com maior conhecimento da linguagem de programação R. Além disso, o painel de resultados é composto por gráficos interativos que facilitam a interpretação. Além disso, aproveitando o crescente ecossistema de serviços web bioinformáticos, nossa ferramenta estabelece uma interface com a plataforma Enrichr (Chen et al., 2013), permitindo uma experiência mais rica para os nossos usuários.
Results
We demonstrated that webCEMiTool can be applied to analyze expression data at the single cell level. Os dados viscRNA-Seq disponíveis ao público (vírus – incluindo ARN de células únicas-Seq) foram obtidos na base de dados GEO NCBI (número de adesão GSE110496) e utilizados como input para a análise. Os dados referem-se à transcriptoma de células individuais de hepatoma humano (Huh7), infectadas quer com o vírus dengue (DENV) quer com o vírus Zika (ZIKV), utilizando multiplicidade de infecções (MOI) 0, 1 ou 10 (Zanini et al., 2018). As células recolhidas em quatro pontos temporais diferentes (4, 12, 24 e 48 h após a infecção) foram então ordenadas para análise transcriptómica de células únicas com um protocolo Smart-seq2 adaptado (Zanini et al., 2018). O conjunto de dados DENV é composto por 933 células infectadas (MOI = 1 ou 10) e 303 controlos (MOI = 0), enquanto o conjunto de dados ZIKV é composto por 488 células infectadas (MOI = 1) e 403 controlos. Antes de submeter a análise à plataforma webCEMiTool, ambos os conjuntos de dados foram transformados log10 e genes que não foram expressos em mais de 80% das amostras foram removidos. Os conjuntos de dados foram então divididos por vírus e por ponto de tempo e usados como entrada (campo”Expression file”) para webCEMiTool. Além dos dados de expressão genética, nós também fornecemos ao webCEMiTool os fenótipos de amostra (i.e., cargas virais) e Reactome gene define.
as nossas análises webCEMiTool geraram uma média de seis módulos por ponto temporal na infecção por DENV e mais de oito módulos por ponto temporal na infecção por ZIKV. Selecionamos um módulo por ponto de tempo como um representante de nossas descobertas (figura 2A). É evidente que, a 24 e 48 horas após a infecção, a actividade de expressão dos módulos representativos aumenta de acordo com a carga viral (figura 2A). Em seguida, realizámos a análise de enriquecimento Via Via dos módulos representativos 24 horas após a infecção, utilizando a ligação webCEMiTool para o Enrichr (figura 2B). Estas conclusões não só corroboram o que foi descrito na publicação original (Zanini et al., 2018), mas também fornecer novas informações sobre a fisiopatologia das infecções virais de dengue e Zika.
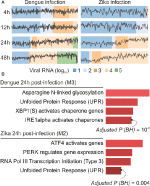
Figura 2. webCEMiTool aplicado a dados RNA-seq unicelulares. A) perfil dos módulos genéticos Co-expressos. Selecionamos um módulo representativo para cada ponto Temporal pós-infecção pelo vírus da dengue (esquerda) ou infecção pelo vírus pós-Zika (direita). A linha preta representa a atividade de expressão mediana dos genes dos módulos em cada amostra. As cores representam a quantidade diferente de RNA do vírus dentro da célula. B) análise da sobre-Representação de módulos seleccionados às 24 horas após a infecção pelo vírus. Os gráficos de barras foram adaptados a partir do Enrichr webtool ligado ao webCEMiTool. As barras são proporcionais ao valor p ajustado-log10 (Benjamini-Hochberg) das vias enriquecidas em um módulo.
discussão
embora poucas aplicações semelhantes baseadas na web tenham sido desenvolvidas para realizar a análise de genes de co-expressão (Tzfadia et al., 2016; Desai et al., 2017), estas ferramentas não fornecem resultados comparáveis ao webCEMiTool. Uma dessas aplicações é GeNET (Desai et al., 2017). Este webtool foi concebido para facilitar as análises de co-expressão genética e fornece análises de enriquecimento e redes gene-a-gene. No entanto, apenas realiza estas análises para três organismos (R. capsulatus, M. tuberculosis, e O. sativa). Outro exemplo é o CoExpNetViz (Tzfadia et al., 2016), um webtool projetado para a visualização e construção de redes de genes. Similar ao GeNET, o CoExpNetViz é um pouco limitado em relação aos organismos, já que se afirma ser principalmente projetado para transcriptomas vegetais. O webCEMiTool tem como objetivo fornecer análises de co-expressão para qualquer organismo. Além disso, embora o CoExpNetViz seja apresentado como uma aplicação baseada na web, seus resultados são devolvidos aos usuários como uma pasta comprimida contendo um README.arquivo txt com instruções sobre como visualizar seus resultados no aplicativo Cytoscape. Os usuários têm então que inserir manualmente na Citoscape os vários arquivos de saída diferentes fornecidos pela ferramenta. Estes passos adicionais também podem tornar o processo propenso a erros e, possivelmente, assustador para os usuários não familiarizados com a Citoscape. O webCEMiTool oferece resultados muito mais convenientes exibidos pelo navegador.
também mostramos que o webCEMitool é capaz de analisar os dados RNA-seq de células únicas de forma mais rápida e eficiente. Nossos resultados devolveram informações relevantes sobre os processos biológicos envolvidos com a infecção pelo vírus dengue e Zika. Toda esta análise foi realizada de forma automatizada e prática, sem necessidade de o usuário ter uma compreensão profunda sobre o processamento interno da análise de dados de co-expressão genética.
contribuições dos autores
LC, PR, BG-C e MA-P realizaram as análises. LC, GS-H E VM-C desenvolveram o webtool. A HN concebeu a ferramenta e supervisionou o trabalho. Todos os autores ajudam na escrita do artigo.
Declaração de conflito de interesses
os autores declaram que a investigação foi realizada na ausência de quaisquer relações comerciais ou financeiras que possam ser interpretadas como um potencial conflito de interesses.