Frontiers in Genetics
Introduction
Cellular processes are driven by multiple interacting molecules whose activity level must be dynamically regulated (Kitano, 2002). Tämän seurauksena geenit, jotka kuuluvat samaan signalointi-ja metaboliareittiin tai joilla on samanlaisia toimintoja, ovat yleensä yhteisilmoitettavissa eri olosuhteissa (Wang et al., 2016). Co-expression-geenimoduulianalyysi luo verkostoja, jotka koostuvat joukoista geenejä (eli moduuleja), joiden ekspressio korreloi voimakkaasti. Tällaista analyysia sovellettiin paljastamaan infektioon liittyviä toiminnallisia moduuleja (Janova et al., 2015), tulehduksellinen (Beins et al., 2016), ja neurologinen (Voineagu et al., 2011) sairaudet sekä useat syöpätyypit (Sharma et al., 2017).
Painotettu geenin co-expression network analysis (WGCNA) on laajalti käytetty menetelmä co-expression-geenimoduulien tunnistamiseksi (Zhang and Horvath, 2005). Wgcna: n suorittaminen edellyttää kuitenkin, että käyttäjät tuntevat ohjelmointiympäristöt sekä valitsevat manuaalisesti parametrit. Nämä ominaisuudet estävät tutkijoita, joilla ei ole riittävää tietoa R: stä, tunnistamasta geenimoduuleja transkriptomitietoaineistoista.
perustuu Biojohdin R-pakettiin nimeltä CEMiTool (Russo et al., 2018), kehitimme käyttäjäystävällisen verkkopohjaisen sovelluksen, jonka avulla tutkijat, joilla ei ole bioinformatiikan taustaa, voivat suorittaa kattavan co-expression-verkkoanalyysin.
materiaalit ja menetelmät
webcemitoolin verkkoliittymä kehitettiin niin, että käyttäjät pystyivät tuottamaan nopeasti kattavia analyysejä ilman, että heidän tarvitsi asentaa mitään tiettyä ohjelmaa tai internet-selainta. Ainoa vaatimus modulaarisen analyysin suorittamiselle on tietojoukko, joka sisältää kaikkien geenien ekspressiotasot näytteissä erilaisissa biologisissa olosuhteissa (tässä määritellään “luokiksi”). Näytteiden vaihteluväliä ei ole määritelty, mutta aiempi tutkimuksemme ehdottaa vähintään 15 näytettä tietokokonaisuutta kohti (Russo et al., 2018). Vaikka se suunniteltiin ensisijaisesti transkriptomidataa varten (eli RNA-seq tai mikroarrayt), sitä voidaan mahdollisesti käyttää myös proteiinien, sytokiinien ja jopa metaboliittien moduulien tunnistamiseen. webCEMiTool valitsee sitten automaattisesti tulogeenit ja tunnistaa co-expression-moduulit. Jokainen moduuli sisältää joukon geenejä, joiden ilmentyminen noudattaa samanlaista kaavaa.
toteutimme webcemitoolissa ominaisuuden, joka arvioi geenimoduulien aktiivisuutta kussakin näyteluokassa. Tätä varten käyttäjien tarvitsee vain antaa näytehuomautus välilehdellä rajattu tekstitiedosto, joka ilmoittaa kunkin näytteen luokan. Tämän jälkeen työkalun” tulokset “- osiossa esitetään” profiilikuva”, joka osoittaa moduulin yksittäisten geenien mediaanitason (Kuva 1a).
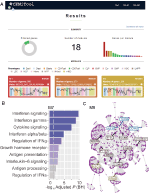
Kuva 1. webCEMiTool yleiskatsaus. (A) webCEMiTool tulokset yhteenveto – donitsi kaavio edustaa osuus valittujen geenien valvomaton suodatin. Etusivulla näkyy myös saatujen moduulien määrä sekä pylväskaavio, joka kuvaa geenien määrää kussakin moduulissa. Moduuliprofiilin käyrät kuvaavat moduulien geenien mediaaniekspressioaktiivisuutta kussakin näytteessä. Värit edustavat eri näyteluokkia. (B) Yliedustuksen analyysi – Tämä kuvaa −log10 adjusted p-arvo (Benjamini-Hochberg) on rikastettu polkuja moduulissa (polut määritellään käyttäjä-inputted .gmt-tiedosto). (C) geeniverkosto moduuli – alkuun eniten kytketty geenit (navat) on merkitty ja värillinen perustuu siihen, ovatko ne alun perin läsnä moduulin (sininen), tai lisätään käyttäjän syötetty vuorovaikutus tiedosto (punainen), tai molemmat (vihreä).
funktionaalisen analyysin mahdollistamiseksi käyttäjät voivat myös tarkistaa, liittyvätkö geenimoduulit tiettyihin signalointi-tai metaboliareitteihin (Kuva 1B). Nämä polut voidaan helposti purkaa tietokannoista, kuten KEGG, Reactome ja MySigDB. Lopuksi käyttäjät voivat integroida tulokset interactome-tietoihin (eli proteiini-proteiini-vuorovaikutuksiin, transkriptiotekijöihin ja niiden transkriboituihin geeneihin tai jopa mirnoihin ja niiden kohdegeeneihin). Tämän ominaisuuden avulla käyttäjät voivat tunnistaa moduulien kriittiset sääntelyviranomaiset (Kuva 1c), mikä antaa arvokasta tietoa kokeellista validointia tai mahdollisia huumekohteita varten. Lisätietoja siitä, miten saada valinnaisia tiedostoja löytyy “opetusohjelma” sivu verkkosivuilla.2
osoittaaksemme, että menetelmämme on kestävä, suoritimme ennennäkemättömän laajan modulaarisen analyysin yli 1000 julkisesti saatavilla olevalla RNA-seq-ja microarray-tietokokonaisuudella sekä uusilla RNA-seq-tiedoilla Leishmaniaan sairastuneista potilaista käyttäen cemitool R-pakkausversiota (Russo et al., 2018). Vaikka webcemitoolissa ja paketissa on erilliset visualisointiominaisuudet ja ne perustuvat eri alustoille, ydinkoeilmaisutoiminnot ovat pääosin samat. Tässä kuvaamamme verkkotyökalu on rakennettu mahdollistamaan helpon pääsyn gene-modulaarisiin analyyseihin ei-ohjelmoiville tutkijoille, kun taas R-kirjastoversio on suunnattu käyttäjille, joilla on enemmän tietoa R – ohjelmointikielestä. Lisäksi tulospaneeli koostuu interaktiivisista kaavioista, jotka helpottavat tulkintaa. Lisäksi hyödyntämällä bioinformatiikan verkkopalveluiden kasvavaa ekosysteemiä, työkalumme muodostaa käyttöliittymän Enrichr-alustan kanssa (Chen et al., 2013), mahdollistaen käyttäjillemme rikkaamman kokemuksen.
tulokset
osoitimme, että webcemitoolia voidaan käyttää analysoimaan ekspressiotietoja yksisolutasolla. Julkisesti saatavilla olevat viskrna-Seq-tiedot (virus-mukaan lukien yksisoluinen RNA-seq) saatiin NCBI: n GEO-tietokannasta (liittymisnumero GSE110496), ja niitä käytettiin analyysissä. Tiedot viittaavat transcriptome yksittäisten ihmisen hepatoma (Huh7) soluja, jotka olivat tartunnan joko denguevirus (DENV) tai zikavirus (ZIKV), käyttäen moninaisuus infektio (MOI) 0, 1, tai 10 (Zanini et al., 2018). Solut, jotka kerättiin neljässä eri aikapisteessä (4, 12, 24 ja 48 h tartunnan jälkeen), lajiteltiin sitten yksisoluista transkriptomianalyysiä varten mukautetulla Smart-seq2-protokollalla (Zanini et al., 2018). DENV-tietokokonaisuus käsittää 933 infektoitunutta solua (MOI = 1 tai 10) ja 303 kontrollia (MOI = 0), kun taas ZIKV-tietokokonaisuus koostuu 488 infektoituneesta solusta (MOI = 1) ja 403 kontrollista. Ennen analyysin toimittamista webCEMiTool-alustalle molemmat tietojoukot muunnettiin log10: ksi ja geenit, joita ei ilmaistu yli 80 prosentissa näytteistä, poistettiin. Tämän jälkeen tietojoukot jaettiin viruksen ja aikapisteen mukaan ja niitä käytettiin syötteenä (“Expression file” – kenttä) webcemitooliin. Geeniekspressiotietojen lisäksi toimitimme webcemitoolille myös näytteen fenotyypit (ts., viruskuormat) ja Reactome-geenijoukot.
webCEMiTool-analyysimme tuottivat keskimäärin kuusi moduulia aikapistettä kohti DENV-tartunnassa ja yli kahdeksan moduulia aikapistettä kohti ZIKV-tartunnassa. Olemme valinneet havaintojemme edustajaksi yhden moduulin aikapistettä kohti (Kuva 2a). On selvää, että 24 ja 48 tunnin kuluttua tartunnasta edustavien moduulien ilmaisuaktiivisuus kasvaa viruskuorman mukaan (kuva 2a). Seuraavaksi suoritimme edustavien moduulien rikastusanalyysin 24 tunnin kuluttua tartunnasta käyttäen webCEMiTool-linkkiä Enrichr: lle (Kuva 2b). Nämä havainnot eivät ainoastaan vahvista sitä, mitä kuvattiin alkuperäisessä julkaisussa (Zanini et al., 2018), mutta tarjoavat myös uusia oivalluksia dengue-ja Zika-virustartuntojen fysiologiasta.
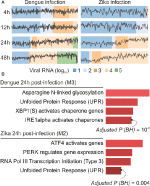
kuva 2. webCEMiTool sovellettu yksisoluinen RNA-seq tiedot. (A) yhdessä ilmaistujen geenimoduulien profiilikuva. Valitsimme yhden edustavan moduulin kullekin aikapisteelle denguevirustartunnan jälkeen (vas.) tai zikavirustartunnan jälkeen (oik.). Musta viiva kuvaa moduulien geenien mediaaniekspressioaktiivisuutta kussakin näytteessä. Värit edustavat viruksen RNA: n eri määrää solun sisällä. B) valittujen moduulien Yliedustusanalyysi 24 tunnin kuluttua virustartunnasta. Pylväsdiagrammit on sovitettu webcemitooliin linkitetystä enrichr webtoolista. Tangot ovat verrannollisia moduulin rikastettujen reittien-log10-mukautettuun p-arvoon (Benjamini-Hochberg).
Keskustelu
vaikka vain harvoja vastaavia web-pohjaisia sovelluksia kehitettiin co-expression-geenianalyysin suorittamiseen (Tzfadia et al., 2016; Desai et al., 2017), nämä työkalut eivät tarjoa vertailukelpoisia tuloksia webCEMiTool. Yksi tällainen sovellus on GeNET (Desai et al., 2017). Tämä webtool on suunniteltu helpottamaan geenien yhteisekspressioanalyysiä ja tarjoaa rikastusanalyysiä ja geenien välisiä verkostoja. Se tekee kuitenkin nämä analyysit vain kolmelle eliölle (R. capsulatus, M. tuberculosis ja O. sativa). Toinen esimerkki on CoExpNetViz (Tzfadia et al., 2016), geeniverkkojen visualisointiin ja rakentamiseen suunniteltu webtool. Genetin tavoin CoExpNetViz on jonkin verran rajoitettu eliöiden suhteen, koska sen on todettu olevan tarkoitettu ensisijaisesti kasvien transkriptomeille. WebCEMiTool pyrkii tarjoamaan rinnakkaisilmaisuanalyysejä mille tahansa organismille. Lisäksi, vaikka CoExpNetViz esitetään verkkopohjaisena sovelluksena, sen tulokset palautetaan käyttäjille pakattuna kansiona, joka sisältää Readme-sovelluksen.txt-tiedosto, jossa on ohjeet tulosten visualisointiin Cytoscape-sovelluksessa. Käyttäjät ovat sitten manuaalisesti lisätä Cytoscape useita eri lähtötiedostoja työkalun. Nämä lisävaiheet voivat myös tehdä prosessista virhealttiin ja mahdollisesti pelottavan käyttäjille, jotka eivät tunne Cytoscapea. WebCEMiTool tarjoaa paljon kätevämpiä selaimen näyttämiä tuloksia.
osoitimme myös, että webCEMitool pystyy analysoimaan yksisoluista RNA-seq-dataa nopeammin ja tehokkaasti. Tuloksemme toivat relevanttia tietoa denguekuume-ja zikavirusinfektion biologisista prosesseista. Kaikki tämä analyysi suoritettiin automatisoidulla ja käytännöllisellä tavalla, eikä käyttäjällä tarvitse olla syvää ymmärrystä geenien co-expression data-analyysin sisäisestä prosessoinnista.
tekijän osuudet
LC, PR, BG-C ja MA-P suorittivat analyysit. LC, GS-H ja VM-C kehittivät webtool-työkalun. HN suunnitteli työkalun ja valvoi työtä. Kaikki kirjoittajat auttavat lehden kirjoittamisessa.
Eturistiriitalausunto
kirjoittajat toteavat, että tutkimus tehtiin ilman kaupallisia tai taloudellisia suhteita, joita voitaisiin pitää mahdollisena eturistiriitana.