Frontiers in Genetics
Introducere
procesele celulare sunt conduse de mai multe molecule care interacționează al căror nivel de activitate trebuie să fie reglat dinamic (Kitano, 2002). Ca urmare, genele care aparțin aceleiași căi de semnalizare și metabolice sau care împărtășesc funcții similare vor tinde să fie co-exprimate între condiții (Wang și colab., 2016). Analiza modulului genetic de coexpresie creează rețele cuprinzând seturi de gene (adică module) a căror expresie este foarte corelată. O astfel de analiză a fost aplicată pentru a dezvălui module funcționale legate de infecțioase (Janova și colab., 2015), inflamator (Beins și colab., 2016) și neurologice (Voineagu și colab., 2011) boli, precum și mai multe tipuri de cancer (Sharma și colab., 2017).
Analiza rețelei de co-exprimare a genelor ponderate (WGCNA) este o metodă utilizată pe scară largă pentru identificarea modulelor de gene co-exprimate (Zhang și Horvath, 2005). Cu toate acestea, pentru a rula WGCNA, utilizatorii trebuie să fie familiarizați cu mediile de programare, precum și să selecteze manual parametrii. Aceste caracteristici împiedică cercetătorii cu cunoștințe insuficiente despre R să identifice modulele genetice din seturile de date transcriptome.
bazat pe pachetul nostru Bioconductor R numit CEMiTool (Russo și colab., 2018), am dezvoltat o aplicație web ușor de utilizat, care permite oamenilor de știință fără experiență în bioinformatică să efectueze o analiză cuprinzătoare a rețelei de co-exprimare.
materiale și metode
interfața web a webCEMiTool a fost dezvoltat pentru a permite utilizatorilor să genereze rapid analize complete, fără a fi nevoie de a instala orice program specific sau browser de internet. Singura cerință pentru rularea analizei modulare este un set de date care conține nivelurile de Expresie ale tuturor genelor din probe în diferite condiții biologice (definite aici ca “clase”). Nu există un număr definit de eșantioane, dar studiul nostru anterior sugerează un minim de 15 eșantioane pe set de date (Russo și colab., 2018). Deși a fost conceput în primul rând pentru datele transcriptomilor (adică ARN-seq sau microarrays), poate fi, de asemenea, utilizat potențial pentru identificarea modulelor de proteine, citokine și chiar metaboliți. webCEMiTool va selecta automat genele de intrare și va identifica modulele de co-exprimare. Fiecare modul conține un set de gene a căror expresie urmează un model similar.
am implementat, în cadrul webCEMiTool, o caracteristică care evaluează activitatea modulelor genetice pe fiecare clasă de probe. Pentru aceasta, utilizatorii trebuie doar să furnizeze un fișier text delimitat de fila de adnotare a eșantionului care informează clasa fiecărui eșantion. Un ” grafic de profil “care arată nivelul median al genelor individuale din cadrul modulului este apoi afișat în secțiunea” Rezultate ” a instrumentului (figura 1a).
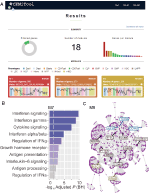
Figura 1. webCEMiTool prezentare generală. (A) rezumatul rezultatelor webCEMiTool – diagrama gogoașă reprezintă proporția genelor selectate de filtrul nesupravegheat. Prima pagină afișează, de asemenea, numărul de module obținute, precum și o diagramă cu bare care prezintă numărul de gene din fiecare modul. Graficele profilului modulului ilustrează activitatea mediană de expresie a genelor din modulele din fiecare eșantion. Culorile reprezintă diferitele clase de probă. (B) analiza Suprareprezentării – aceasta descrie valoarea p ajustată −log10 (Benjamin-Hochberg) a căilor îmbogățite într-un modul (căi definite de utilizatorul introdus .fișier gmt). (C) rețeaua genetică a unui modul – cele mai conectate gene (hub-uri) sunt etichetate și colorate în funcție de faptul dacă au fost inițial prezente în modul (albastru) sau inserate dintr-un fișier de interacțiune introdus de utilizator (roșu) sau ambele (verde).
pentru a permite analiza funcțională, utilizatorii pot verifica, de asemenea, dacă modulele genetice sunt asociate cu căi specifice de semnalizare sau metabolice (figura 1b). Aceste căi pot fi extrase cu ușurință din baze de date, cum ar fi KEGG, Reactome și MySigDB. În cele din urmă, utilizatorii pot integra rezultatele cu date interacomice (adică interacțiuni proteine-proteine, factori de transcripție și genele lor transcrise sau chiar miARN și genele lor țintă). Această caracteristică permite utilizatorilor să identifice regulatorii critici ai modulelor (figura 1C), oferind informații valoroase pentru validarea experimentală sau ținte potențiale pentru medicamente. Detalii suplimentare despre modul de obținere a fișierelor opționale pot fi găsite în pagina “Tutorial” a site-ului web.2
pentru a demonstra că metoda noastră este robustă, am efectuat o analiză modulară la scară largă fără precedent, cu peste 1.000 de seturi de date ARN-seq și microarray disponibile publicului și noi date ARN-seq ale pacienților infectați cu Leishmania folosind versiunea pachetului cemitool R (Russo și colab., 2018). Deși webCEMiTool și pachetul au caracteristici distincte de vizualizare și se bazează pe platforme diferite, funcționalitatea de bază a co-expresiei este în esență aceeași. Instrumentul online pe care îl descriem aici este construit pentru a permite accesul ușor la analizele modulare genetice pentru cercetătorii care nu programează, în timp ce versiunea R library este orientată către utilizatorii cu o mai mare cunoaștere a limbajului de programare R. În plus, tabloul de bord rezultate este compus din diagrame interactive care facilitează interpretarea. Mai mult, profitând de ecosistemul în creștere al serviciilor web bioinformatice, instrumentul nostru stabilește o interfață cu platforma Enrichr (Chen și colab., 2013), permițând o experiență mai bogată pentru utilizatorii noștri.
rezultate
am demonstrat că webCEMiTool poate fi aplicat pentru a analiza datele de Expresie la nivel de celulă unică. Datele viscRNA-Seq disponibile publicului (virus-inclusiv ARN cu o singură celulă-Seq) au fost obținute din Baza de date geo NCBI (numărul de aderare GSE110496) și utilizate ca intrare pentru analiză. Datele se referă la transcriptomul celulelor individuale ale hepatomului uman (Huh7), care au fost infectate fie cu virusul dengue (DENV), fie cu virusul Zika (ZIKV), folosind multiplicitatea infecției (MOI) 0, 1 sau 10 (Zanini și colab., 2018). Celulele colectate pe patru puncte de timp diferite (4, 12, 24 și 48 de ore după infecție) au fost apoi sortate pentru analiza transcriptomică cu o singură celulă cu un protocol Smart-seq2 adaptat (Zanini și colab., 2018). Setul de date DENV cuprinde 933 de celule infectate (MOI = 1 sau 10) și 303 de controale (MOI = 0), în timp ce setul de date ZIKV este compus din 488 de celule infectate (MOI = 1) și 403 de controale. Înainte de a trimite analiza pe platforma webCEMiTool, ambele seturi de date au fost transformate log10 și genele care nu au fost exprimate în mai mult de 80% din probe au fost eliminate. Seturile de date au fost apoi împărțite în funcție de virus și de punctul de timp și utilizate ca intrare (câmpul”Expression file”) la webCEMiTool. În plus față de datele privind expresia genelor, am furnizat, de asemenea, webCEMiTool fenotipurile eșantionului (adică., încărcături virale) și seturi de gene Reactome.
analizele noastre webCEMiTool au generat în medie șase module pe punct de timp în infecția cu DENV și mai mult de opt module pe punct de timp în infecția cu ZIKV. Am selectat un modul pentru fiecare punct de timp ca reprezentant al constatărilor noastre (figura 2A). Este clar că la 24 și 48 de ore post-infecție, activitatea de expresie a modulelor reprezentative crește în funcție de încărcătura virală (figura 2a). În continuare am efectuat analiza de îmbogățire pe cale a modulelor reprezentative la 24 h post-infecție utilizând link-ul webCEMiTool pentru Enrichr (figura 2b). Aceste constatări nu numai că confirmă ceea ce a fost descris în publicația originală (Zanini și colab., 2018), dar oferă, de asemenea, noi informații despre fiziopatologia infecțiilor cu virusul dengue și Zika.
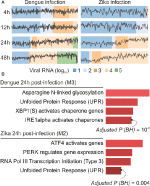
Figura 2. webCEMiTool aplicat datelor ARN-seq cu o singură celulă. (A) graficul profilului modulelor genetice coexprimate. Am selectat un modul reprezentativ pentru fiecare punct de timp după infecția cu virusul dengue (stânga) sau după infecția cu virusul Zika (dreapta). Linia neagră reprezintă activitatea mediană de expresie a genelor din modulele din fiecare probă. Culorile reprezintă cantitatea diferită de ARN de virus din celulă. (B) analiza Suprareprezentării modulelor selectate la 24 de ore după infecția cu virus. Graficele cu bare au fost adaptate de la enrichr webtool legat de webCEMiTool. Barele sunt proporționale cu valoarea p ajustată-log10 (Benjamin-Hochberg) a căilor îmbogățite într-un modul.
discuție
deși puține aplicații similare bazate pe web au fost dezvoltate pentru a efectua analiza genelor de coexpresie (Tzfadia și colab., 2016; Desai și colab., 2017), aceste instrumente nu oferă rezultate comparabile cu webCEMiTool. O astfel de aplicație este GeNET (Desai și colab., 2017). Acest webtool a fost conceput pentru a facilita analizele de co-exprimare a genelor și oferă analize de îmbogățire și rețele gene-to-gene. Cu toate acestea, efectuează aceste analize numai pentru trei organisme (R. capsulatus, M. tuberculosis și O. sativa). Un alt exemplu este CoExpNetViz (Tzfadia și colab., 2016), un instrument web conceput pentru vizualizarea și construirea rețelelor genetice. Similar cu GeNET, CoExpNetViz este oarecum limitat în ceea ce privește organismele, deoarece se afirmă că este conceput în primul rând pentru transcriptomii plantelor. WebCEMiTool își propune să ofere analize de co-exprimare pentru orice organism. Mai mult, deși CoExpNetViz este prezentat ca o aplicație bazată pe web, rezultatele sale sunt returnate utilizatorilor ca un folder comprimat care conține un README.fișier txt cu instrucțiuni despre cum să vizualizați rezultatele lor în aplicația Cytoscape. Utilizatorii trebuie apoi să introducă manual în Cytoscape mai multe fișiere de ieșire diferite furnizate de instrument. Acești pași suplimentari pot face, de asemenea, procesul predispus la erori și, eventual, descurajant pentru utilizatorii care nu sunt familiarizați cu Cytoscape. WebCEMiTool oferă rezultate mult mai convenabile afișate în browser.
am arătat, de asemenea, că webCEMitool este capabil să analizeze datele ARN-seq cu o singură celulă mai rapid și mai eficient. Rezultatele noastre au returnat informații relevante despre procesele biologice implicate în infecția cu dengue și virusul Zika. Toate aceste analize au fost efectuate într-o manieră automată și practică, fără a fi nevoie ca utilizatorul să aibă o înțelegere profundă asupra procesării interne a analizei datelor de coexpresie genetică.
contribuțiile autorului
LC, PR, BG-C și MA-P au efectuat analizele. LC, GS-H și VM-C au dezvoltat instrumentul webtool. HN a conceput instrumentul și a supravegheat lucrarea. Toți autorii ajută la scrierea lucrării.
Declarație privind conflictul de interese
autorii declară că cercetarea a fost realizată în absența oricăror relații comerciale sau financiare care ar putea fi interpretate ca un potențial conflict de interese.