Frontiers in Genetics
Bevezetés
a sejtes folyamatokat több kölcsönható molekula vezérli, amelyek aktivitási szintjét dinamikusan szabályozni kell (Kitano, 2002). Ennek eredményeként az azonos jelátviteli és metabolikus útvonalhoz tartozó vagy hasonló funkciókat megosztó gének általában együtt expresszálódnak a körülmények között (Wang et al., 2016). A Ko-expressziós génmodul-elemzés olyan génkészletekből (azaz modulokból) álló hálózatokat hoz létre, amelyek expressziója erősen korrelál. Ilyen elemzést alkalmaztak a fertőzéssel kapcsolatos funkcionális modulok feltárására (Janova et al., 2015), gyulladásos (Beins et al., 2016) és neurológiai (Voineagu et al., 2011) betegségek, valamint többféle rák (Sharma et al., 2017).
a Weighted gene co-expression network analysis (Wgcna) egy széles körben használt módszer a co-expresszált génmodulok azonosítására (Zhang and Horvath, 2005). A WGCNA futtatásához azonban a felhasználóknak ismerniük kell a programozási környezeteket, valamint manuálisan kell kiválasztaniuk a paramétereket. Ezek a tulajdonságok megakadályozzák a kutatókat, akiknek nincs elegendő ismerete az R-ről a génmodulok azonosítására a transzkriptóm adatkészletekből.
a Cemitool nevű Bioconductor r csomag alapján (Russo et al., 2018), kifejlesztettünk egy felhasználóbarát webalapú alkalmazást, amely lehetővé teszi a bioinformatikai háttérrel nem rendelkező tudósok számára, hogy átfogó co-expression hálózati elemzést végezzenek.
anyagok és módszerek
a webCEMiTool webes felületét úgy fejlesztették ki, hogy lehetővé tegye a felhasználók számára, hogy gyorsan átfogó elemzéseket készítsenek anélkül, hogy bármilyen speciális programot vagy internetböngészőt kellene telepíteniük. A moduláris elemzés futtatásának egyetlen követelménye egy olyan adatkészlet, amely a mintákban lévő összes gén expressziós szintjét tartalmazza különböző biológiai körülmények között (a továbbiakban “osztályok”). Nincs meghatározott tartományú Minták száma, de korábbi tanulmányunk adatkészletenként legalább 15 mintát javasol (Russo et al., 2018). Bár elsősorban transzkriptóm adatokra tervezték (azaz RNS-seq vagy mikroarray), potenciálisan felhasználható fehérjék, citokinek, sőt metabolitok moduljainak azonosítására is. a webCEMiTool ezután automatikusan kiválasztja a bemeneti géneket, és azonosítja a Ko-expressziós modulokat. Minden modul tartalmaz egy génkészletet, amelynek expressziója hasonló mintát követ.
a webCEMiTool-on belül megvalósítottunk egy olyan funkciót, amely értékeli a génmodulok aktivitását az egyes mintaosztályokon. Ehhez a felhasználóknak csak egy minta annotációs tabulátorral tagolt szövegfájlt kell megadniuk, amely tájékoztatja az egyes minták osztályát. Ezután az eszköz” eredmények “részében megjelenik egy” profilábra”, amely a modulon belüli egyes gének medián szintjét mutatja (1a ábra).
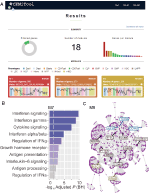
1. ábra. webCEMiTool áttekintés. (A) webCEMiTool eredmények összefoglalása – a fánk diagram a kiválasztott gének arányát mutatja a felügyelet nélküli szűrő által. Az első oldalon megjelenik a kapott modulok száma, valamint egy oszlopdiagram, amely az egyes modulok génjeinek számát ábrázolja. A modulprofil diagramok szemléltetik a modulok génjeinek medián expressziós aktivitását az egyes mintákban. A színek a különböző mintaosztályokat képviselik. (B) Felülreprezentációs elemzés – ez ábrázolja a −log10 korrigált p-érték (Benjamini-Hochberg) a dúsított utak egy modulban (utak által meghatározott felhasználó által bevitt .gmt fájl). (C) egy modul Génhálózata – a legjobban összekapcsolt géneket (hubok) az alapján jelölik és színezik, hogy eredetileg jelen voltak-e a modulban (kék), vagy egy felhasználó által bevitt interakciós fájlból (piros) vagy mindkettőből (zöld) kerültek-e be.
a funkcionális elemzés lehetővé tétele érdekében a felhasználók azt is ellenőrizhetik, hogy a génmodulok specifikus jelátviteli vagy metabolikus útvonalakhoz kapcsolódnak-e (1b ábra). Ezek az útvonalak könnyen kinyerhetők olyan adatbázisokból, mint a KEGG, a Reactome és a MySigDB. Végül a felhasználók integrálhatják az eredményeket az interaktom adatokkal (azaz a fehérje-fehérje kölcsönhatásokkal, a transzkripciós faktorokkal és az átírt génjeikkel, vagy akár a miRNS-ekkel és a célgénjeikkel). Ez a funkció lehetővé teszi a felhasználók számára a modulok kritikus szabályozóinak azonosítását (1C ábra), értékes betekintést nyújtva a kísérleti validációhoz vagy a gyógyszerek potenciális célpontjaihoz. Az opcionális fájlok beszerzésével kapcsolatos további részletek a weboldal “bemutató” oldalán találhatók.2
annak bizonyítására, hogy módszerünk robusztus, példátlan nagyszabású moduláris elemzést végeztünk több mint 1000 nyilvánosan elérhető RNS-seq és microarray adatkészlettel, valamint a Leishmania-val fertőzött betegek új RNS-seq adataival a CEMiTool r csomag verziójával (Russo et al., 2018). Bár a webCEMiTool és a csomag különböző vizualizációs funkciókkal rendelkezik, és különböző platformokon alapulnak, az alapvető co-expression funkció lényegében ugyanaz. Az itt leírt online eszköz úgy van kialakítva, hogy a nem programozási kutatók számára könnyű hozzáférést biztosítson a génmoduláris elemzésekhez, míg az R könyvtár verziója az R programozási nyelv nagyobb ismeretével rendelkező felhasználók számára készült. Ezenkívül az eredmények irányítópultja interaktív diagramokból áll, amelyek megkönnyítik az értelmezést. Sőt, kihasználva a bioinformatikai webszolgáltatások növekvő ökoszisztémáját, eszközünk interfészt hoz létre az Enrichr platformmal (Chen et al., 2013), gazdagabb élményt nyújtva felhasználóink számára.
eredmények
megmutattuk, hogy a webCEMiTool alkalmazható az expressziós adatok elemzésére egyetlen sejt szintjén. A nyilvánosan elérhető viscrns-Seq adatokat (vírus-beleértve az egysejtű RNS-Seq-t is) az NCBI GEO adatbázisából (csatlakozási szám: GSE110496) nyerték, és inputként használták fel az elemzéshez. Az adatok az egyes humán hepatoma (Huh7) sejtek transzkriptómájára vonatkoznak, amelyek dengue-vírussal (DENV) vagy Zika-vírussal (ZIKV) fertőzöttek, többszörös fertőzés (MOI) felhasználásával 0, 1 vagy 10 (Zanini et al., 2018). A négy különböző időpontban gyűjtött sejteket (4, 12, 24 és 48 órával a fertőzés után) egysejtű transzkriptomikus elemzésre válogattuk egy adaptált Smart-seq2 protokollal (Zanini et al., 2018). A DENV adathalmaz 933 fertőzött sejtből (MOI = 1 vagy 10) és 303 kontrollból (MOI = 0) áll, míg a ZIKV adathalmaz 488 fertőzött sejtből (MOI = 1) és 403 kontrollból áll. Az elemzés webCEMiTool platformra történő benyújtása előtt mindkét adatkészletet log10 transzformáltuk, és azokat a géneket, amelyek a minták több mint 80% – ában nem expresszálódtak ki, eltávolítottuk. Az adathalmazokat ezután vírus és időpont szerint osztották fel, és bemenetként (“Expression file” mező) használták a webCEMiTool-hoz. A génexpressziós adatok mellett a webCEMiTool számára a minta fenotípusait (pl., vírusterhelés) és a Reactome génkészletek.
webCEMiTool elemzéseink átlagosan hat modult generáltak a DENV fertőzés idején, és több mint nyolc modult a ZIKV fertőzés idején. Megállapításaink képviselőjeként időpontonként egy modult választottunk ki (2a.ábra). Nyilvánvaló, hogy a fertőzés utáni 24 és 48 órában a reprezentatív modulok expressziós aktivitása a vírusterhelésnek megfelelően növekszik (2a ábra). Ezután elvégeztük a reprezentatív modulok útvonal-dúsítási elemzését 24 órával a fertőzés után a Webcemitool link segítségével az Enrichr-hez (2b ábra). Ezek a megállapítások nemcsak megerősítik az eredeti kiadványban leírtakat (Zanini et al., 2018), de új betekintést nyújt a dengue-és Zika-vírusfertőzések fiziopatológiájába is.
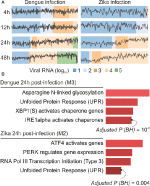
2. ábra. webCEMiTool alkalmazott egysejtű RNS-seq adatok. A) az Együtt expresszált génmodulok profildiagramja. Kiválasztottunk egy reprezentatív modult minden időpontra a dengue vírusfertőzés után (balra) vagy a Zika vírusfertőzés után (jobbra). A fekete vonal a modulok génjeinek medián expressziós aktivitását képviseli az egyes mintákban. A színek a vírus RNS különböző mennyiségét képviselik a sejten belül. B) a kiválasztott modulok Felülreprezentációs elemzése a vírusfertőzés utáni 24 órában. Az oszlopdiagramokat a Webcemitoolhoz kapcsolódó Enrichr webtool-ból adaptáltuk. A rudak arányosak a-log10 korrigált p-érték (Benjamini-Hochberg) a dúsított utak egy modulban.
Vita
bár kevés hasonló webalapú alkalmazást fejlesztettek ki a Ko-expressziós génelemzés elvégzésére (Tzfadia et al., 2016; Desai et al., 2017), ezek az eszközök nem nyújtanak összehasonlítható eredményeket a webCEMiTool-hoz. Az egyik ilyen alkalmazás a GeNET (Desai et al., 2017). Ezt a webeszközt úgy tervezték, hogy megkönnyítse a gén együttexpressziós analíziseket, valamint dúsítási elemzést és gén-gén hálózatokat biztosít. Ezeket az elemzéseket azonban csak három organizmusra (R. capsulatus, M. tuberculosis és O. sativa) végzi. Egy másik példa a CoExpNetViz (Tzfadia et al., 2016), a génhálózatok vizualizálására és felépítésére tervezett webeszköz. A Genethez hasonlóan a CoExpNetViz némileg korlátozott az organizmusok tekintetében, mivel azt állítják, hogy elsősorban növényi transzkriptómákra tervezték. A webCEMiTool célja, hogy Társ-expressziós elemzéseket nyújtson bármely szervezet számára. Sőt, bár CoExpNetViz kerül bemutatásra, mint egy web – alapú alkalmazás, az eredményeket vissza a felhasználóknak, mint egy tömörített mappát tartalmazó README.txt fájl utasításokkal az eredmények megjelenítésére a Cytoscape alkalmazásban. A felhasználóknak ezután manuálisan be kell illeszteniük a Cytoscape-be az eszköz által biztosított különböző kimeneti fájlokat. Ezek a további lépések is, hogy a folyamat hibára hajlamos, és esetleg ijesztő, hogy a felhasználók nem ismerik Cytoscape. A webCEMiTool sokkal kényelmesebb böngészővel megjelenített eredményeket kínál.
azt is megmutattuk, hogy a webCEMitool képes az egysejtű RNS-seq adatok gyorsabb és hatékonyabb elemzésére. Eredményeink releváns információkat szolgáltattak a dengue-és Zika-vírusfertőzéssel kapcsolatos biológiai folyamatokról. Mindezeket az elemzéseket automatizált és gyakorlati módon végezték el, anélkül, hogy a felhasználónak mélyen meg kellett volna értenie a gén-expressziós adatok elemzésének belső feldolgozását.
szerzői hozzájárulások
LC, PR, BG-C és MA-P végezte el az elemzéseket. Az LC, a GS-H és a VM-C fejlesztette ki a webtoolt. A HN megtervezte az eszközt és felügyelte a munkát. Minden szerző segít a papír írásában.
összeférhetetlenségi nyilatkozat
a szerzők kijelentik, hogy a kutatást olyan kereskedelmi vagy pénzügyi kapcsolatok hiányában végezték, amelyek potenciális összeférhetetlenségnek tekinthetők.