Frontiers in Genetics
introduktion
cellulära processer drivs av flera interagerande molekyler vars aktivitetsnivå måste regleras dynamiskt (Kitano, 2002). Som ett resultat tenderar gener som tillhör samma signalering och metaboliska väg eller delar liknande funktioner att vara samuttryckta över förhållanden (Wang et al., 2016). Co-expression genmodul analys skapar nätverk innefattande uppsättningar av gener (dvs. moduler) vars uttryck är starkt korrelerad. Sådan analys tillämpades för att avslöja funktionella moduler relaterade till infektiös (Janova et al., 2015), inflammatorisk (Beins et al., 2016) och neurologiska (Voineagu et al., 2011) sjukdomar, liksom flera typer av cancer (Sharma et al., 2017).
Weighted gene co-expression network analysis (WGCNA) är en allmänt använd metod för att identifiera kouttryckta genmoduler (Zhang och Horvath, 2005). För att kunna köra WGCNA måste användarna dock vara bekanta med programmeringsmiljöer samt att manuellt välja parametrar. Dessa funktioner förhindrar forskare med otillräcklig kunskap om R för att identifiera genmoduler från transkriptomdataset.
baserat på vårt Bioconductor R-paket med namnet CEMiTool (Russo et al., 2018) utvecklade vi en användarvänlig webbaserad applikation som gör det möjligt för forskare utan bakgrund inom bioinformatik att utföra omfattande co-expression nätverksanalys.
material och metoder
webbgränssnittet för webCEMiTool utvecklades för att tillåta användare att snabbt generera omfattande analyser utan att behöva installera något specifikt program eller webbläsare. Det enda kravet för att köra den modulära analysen är en datamängd som innehåller uttrycksnivåerna för alla gener i prover under olika biologiska förhållanden (här definieras som “klasser”). Det finns inget definierat intervall antal prover men vår tidigare studie antyder minst 15 prover per datamängd (Russo et al., 2018). Även om det främst var utformat för transkriptomdata (dvs RNA-seq eller mikroarrayer), kan det också potentiellt användas för att identifiera moduler av proteiner, cytokiner och till och med metaboliter. webCEMiTool väljer sedan automatiskt inmatningsgenerna och identifierar samuttrycksmodulerna. Varje modul innehåller en uppsättning gener vars uttryck följer ett liknande mönster.
vi implementerade, inom webCEMiTool, en funktion som bedömer aktiviteten hos genmoduler på varje klass av prover. För detta behöver användarna bara tillhandahålla en tabbavgränsad textfil för exempelannotering som informerar klassen för varje prov. En ” profilplott “som visar mediannivån för enskilda gener inom modulen visas sedan i avsnittet” Resultat ” i verktyget (Figur 1a).
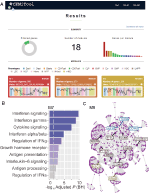
Figur 1. webCEMiTool översikt. (A) webCEMiTool – resultatsammanfattning-munkdiagrammet representerar andelen utvalda gener av det oövervakade filtret. Framsidan visar också antalet erhållna moduler, liksom ett stapeldiagram som visar antalet gener i varje modul. Modulprofildiagram illustrerar medianuttrycksaktiviteten hos gener från Modulerna över varje prov. Färgerna representerar de olika provklasserna. (B) Överrepresentationsanalys – detta visar −log10 justerat p-värde (Benjamini-Hochberg) av de berikade vägarna i en modul (vägar definierade av användarinmatade .GMT-fil). (C) Gennätverk av en modul – de mest anslutna generna (nav) är märkta och färgade baserat på om de ursprungligen var närvarande i modulen (blå) eller infogade från en användarinmatad interaktionsfil (röd) eller båda (grön).
för att möjliggöra funktionell analys kan användarna också kontrollera om genmodulerna är associerade med specifika signalerings-eller metaboliska vägar (Figur 1b). Dessa vägar kan enkelt extraheras från databaser, såsom KEGG, Reactome och MySigDB. Slutligen kan användare integrera resultaten med interaktomdata (dvs. protein-proteininteraktioner, transkriptionsfaktorer och deras transkriberade gener, eller till och med miRNAs och deras målgener). Denna funktion gör det möjligt för användare att identifiera kritiska regulatorer av moduler (figur 1C), vilket ger värdefulla insikter för experimentell validering eller potentiella mål för droger. Ytterligare information om hur du får de valfria filerna finns på sidan “handledning” på webbplatsen.2
för att visa att vår metod är robust utförde vi en aldrig tidigare skådad storskalig modulär analys med över 1000 offentligt tillgängliga RNA-seq-och microarray-dataset och nya RNA-seq-data från patienter infekterade med Leishmania med cemitool r-paketversionen (Russo et al., 2018). Även om webCEMiTool och paketet har distinkta visualiseringsfunktioner och är baserade på olika plattformar, är kärnan co-expression funktionalitet i huvudsak densamma. Onlineverktyget vi beskriver här är byggt för att möjliggöra enkel åtkomst till genmodulära analyser för icke-programmerande forskare, medan R library-versionen är inriktad på användare med större kunskap om r-programmeringsspråket. Dessutom består resultatpanelen av interaktiva diagram som underlättar Tolkning. Dessutom utnyttjar det stigande ekosystemet för bioinformatik webbtjänster, vårt verktyg etablerar ett gränssnitt med Enrichr-plattformen (Chen et al., 2013), vilket möjliggör en rikare upplevelse för våra användare.
resultat
vi visade att webCEMiTool kan användas för att analysera uttrycksdata på encellsnivå. Offentligt tillgängliga viscRNA – seq-data (virus-inklusive Single cell RNA-Seq) erhölls från NCBI GEO database (anslutningsnummer GSE110496) och användes som indata för analysen. Uppgifterna hänvisar till transkriptom av enskilda humana hepatomceller (Huh7), som infekterades med antingen denguevirus (DENV) eller Zika-virus (ZIKV), med användning av multiplicitet av infektion (MOI) 0, 1 eller 10 (Zanini et al., 2018). Celler samlade på fyra olika tidpunkter (4, 12, 24 och 48 timmar efter infektion) sorterades sedan för enkelcellstranskriptomisk analys med ett anpassat Smart-seq2-protokoll (Zanini et al., 2018). Denv-datamängden består av 933 infekterade celler (MOI = 1 eller 10) och 303 kontroller (MOI = 0), medan ZIKV-datamängden består av 488 infekterade celler (MOI = 1) och 403 kontroller. Innan analysen skickades in till webCEMiTool-plattformen transformerades båda datamängderna log10 och gener som inte uttrycktes i mer än 80% av proverna avlägsnades. Datamängderna delades sedan av virus och vid tidpunkt och användes som inmatning (“Expression file” – fält) till webCEMiTool. Förutom genuttrycksdata tillhandahöll vi också till webCEMiTool provfenotyperna (dvs., virala belastningar) och Reaktomgenuppsättningar.
våra webCEMiTool-analyser genererade i genomsnitt sex moduler per tidpunkt i denv-infektion och mer än åtta moduler per tidpunkt i ZIKV-infektion. Vi har valt en modul per tidpunkt som representant för våra resultat (figur 2a). Det är uppenbart att vid 24 och 48 timmar efter infektion ökar expressionsaktiviteten hos representativa moduler beroende på virusbelastningen (figur 2a). Vi utförde därefter analysen av väganrikning av de representativa modulerna vid 24 h efter infektion med webCEMiTool-länken för Enrichr (Figur 2B). Dessa resultat bekräftar inte bara vad som beskrivs i den ursprungliga publikationen (Zanini et al., 2018) men ger också nya insikter om fysiopatologin för dengue-och Zika-virusinfektioner.
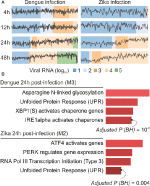
Figur 2. webCEMiTool appliceras på encelliga RNA-seq-data. (A) Profil plot av co-uttryckta genmoduler. Vi valde en representativ modul för varje tidpunkt post-dengue virusinfektion (vänster) eller Post-Zika virusinfektion (höger). Den svarta linjen representerar medianuttrycksaktiviteten hos gener från Modulerna över varje prov. Färgerna representerar den olika mängden virus-RNA i cellen. (B) överrepresentation analys av utvalda moduler vid 24 timmar efter virusinfektion. Stapeldiagrammen anpassades från Enrichr webtool kopplad till webCEMiTool. Staplarna är proportionella mot-log10 justerat p-värde (Benjamini-Hochberg) av de berikade vägarna i en modul.
diskussion
även om få liknande webbaserade applikationer utvecklades för att utföra co-expression genanalys (Tzfadia et al., 2016; Desai et al., 2017) ger dessa verktyg inte jämförbara resultat med webCEMiTool. En sådan ansökan är GeNET (Desai et al., 2017). Denna webtool var utformad för att underlätta gen co-expression analyser och ger anrikning analys och gen-till-Gen nätverk. Det utför emellertid endast dessa analyser för tre organismer (R. capsulatus, M. tuberculosis och O. sativa). Ett annat exempel är CoExpNetViz (Tzfadia et al., 2016), ett webbverktyg utformat för visualisering och konstruktion av gennätverk. I likhet med GeNET är CoExpNetViz något begränsat med avseende på organismerna eftersom det sägs vara främst utformat för växttranskriptomer. WebCEMiTool syftar till att ge co-expression analyser för någon organism. Dessutom, även om CoExpNetViz presenteras som en webbaserad applikation, returneras dess resultat till användare som en komprimerad mapp som innehåller en README.txt-fil med instruktioner om hur man visualiserar sina resultat i Cytoscape-appen. Användarna måste sedan manuellt infoga i Cytoscape de flera olika utdatafilerna som tillhandahålls av verktyget. Dessa ytterligare steg kan också göra processen felbenägen och eventuellt skrämmande för användare som inte känner till Cytoscape. WebCEMiTool erbjuder mycket bekvämare resultat som visas i webbläsaren.
vi visade också att webCEMitool kan analysera encelliga RNA-seq-data snabbare och effektivt. Våra resultat gav relevant information om de biologiska processerna som är involverade i dengue-och Zika-virusinfektion. All denna analys utfördes på ett automatiserat och praktiskt sätt, utan behov av användaren att ha djup förståelse för den interna behandlingen av Gen co-expression dataanalys.
Författarbidrag
LC, PR, BG-C och MA-P utförde analyserna. LC, gs-H och VM-C utvecklade webbverktyget. HN tänkte verktyget och övervakade arbetet. Alla författare hjälper till att skriva papperet.
intressekonflikt uttalande
författarna förklarar att forskningen genomfördes i avsaknad av kommersiella eller finansiella relationer som kan tolkas som en potentiell intressekonflikt.