Frontiers in Genetics
introduktion
cellulære processer drives af flere interagerende molekyler, hvis aktivitetsniveau skal reguleres dynamisk (Kitano, 2002). Som et resultat vil gener, der hører til den samme signalerings-og metaboliske vej eller deler lignende funktioner, have tendens til at blive udtrykt på tværs af betingelser., 2016). Co-ekspressionsgenmodulanalyse skaber netværk bestående af sæt gener (dvs.moduler), hvis ekspression er stærkt korreleret. En sådan analyse blev anvendt til at afsløre funktionelle moduler relateret til infektiøs (Janova et al., 2015), inflammatorisk (Beins et al., 2016) og neurologisk (Voineagu et al., 2011) sygdomme samt flere typer kræft (Sharma et al., 2017).
vægtet gen-Co-ekspressionsnetværksanalyse er en meget anvendt metode til at identificere co-udtrykte genmoduler (hang og Horvath, 2005). For at kunne køre VVCNA skal brugerne dog være bekendt med programmeringsmiljøer samt manuelt vælge parametre. Disse funktioner forhindrer forskere med utilstrækkelig viden om R til at identificere genmoduler fra transkriptomdatasæt.
baseret på vores Bioconductor r-pakke med navnet CEMiTool (Russo et al., 2018), udviklede vi en brugervenlig internetbaseret applikation, der gør det muligt for forskere uden baggrund i bioinformatik at udføre omfattende Co-ekspressionsnetværksanalyse.
materialer og metoder
internetgrænsefladen blev udviklet for at give brugerne mulighed for hurtigt at generere omfattende analyser uden behov for at installere noget specifikt program eller internet. Det eneste krav til at køre den modulære analyse er et datasæt, der indeholder ekspressionsniveauerne for alle gener i prøver under forskellige biologiske betingelser (heri defineret som “klasser”). Der er ikke noget defineret antal prøver, men vores tidligere undersøgelse antyder mindst 15 prøver pr.datasæt (Russo et al., 2018). Selvom det primært var designet til transkriptomdata (dvs.RNA-sekv eller mikroarrays), kan det også potentielt bruges til at identificere moduler af proteiner, cytokiner og endda metabolitter. derefter vælger vi automatisk inputgenerne og identificerer Co-ekspressionsmodulerne. Hvert modul indeholder et sæt gener, hvis udtryk følger et lignende mønster.
Vi implementerede en funktion, der vurderer aktiviteten af genmoduler på hver klasse af prøver. Til dette skal brugerne kun angive en prøveanmærkningsfanebegrænset tekstfil, der informerer klassen for hver prøve. Et “profilplot”, der viser medianniveauet for individuelle gener i modulet, vises derefter i afsnittet” Resultater ” i værktøjet (figur 1a).
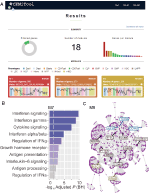
Figur 1. oversigt over emitool. Doughnut-diagrammet repræsenterer andelen af udvalgte gener af det ikke-overvågede filter. Forsiden viser også antallet af opnåede moduler samt et søjlediagram, der viser antallet af gener i hvert modul. Modulprofilplotter illustrerer medianekspressionsaktiviteten af gener fra modulerne på tværs af hver prøve. Farverne repræsenterer de forskellige prøveklasser. (B) overrepræsentationsanalyse – dette viser −log10-justeret p-værdi (Benjamini-Hochberg) af de berigede veje i et modul (veje defineret af brugerinputtet .GMT-fil). (C) Gen netværk af et modul – de øverste mest tilsluttede gener (hubs) er mærket og farvet baseret på, om de oprindeligt var til stede i modulet (blå) eller indsat fra en brugerinputtet interaktionsfil (rød) eller begge dele (grøn).
for at muliggøre funktionel analyse kan brugerne også kontrollere, om genmodulerne er forbundet med specifikke signal-eller metaboliske veje (figur 1b). Disse veje kan let udvindes fra databaser, såsom KEGG, Reactome og MySigDB. Endelig kan brugerne integrere resultaterne med interaktomdata (dvs.protein-protein-interaktioner, transkriptionsfaktorer og deres transkriberede gener eller endda miRNA ‘ er og deres målgener). Denne funktion gør det muligt for brugerne at identificere kritiske regulatorer af moduler (figur 1C), hvilket giver værdifuld indsigt til eksperimentel validering eller potentielle mål for lægemidler. Yderligere oplysninger om, hvordan du får de valgfrie filer kan findes i “Tutorial” side af hjemmesiden.2
for at demonstrere, at vores metode er robust, udførte vi en hidtil uset storskala modulanalyse med over 1.000 offentligt tilgængelige RNA-sekv-og mikroarray-datasæt og nye RNA-sekv-data fra patienter inficeret med Leishmania ved hjælp af CEMiTool r-pakkeversionen (Russo et al., 2018). Selvom
resultater
vi demonstrerede, at cemitool kan anvendes til at analysere ekspressionsdata på enkeltcelleniveau. Offentligt tilgængelige viscRNA-sek-data (virus-inklusive enkeltcelle-RNA-sek) blev opnået fra NCBI GEO-database (tiltrædelsesnummer GSE110496) og anvendt som input til analysen. Dataene henviser til transkriptomet af individuelle humane hepatomceller (Huh7), som var inficeret med enten dengue-virus (Denv) eller Sika-virus (SIKV) ved hjælp af multiplicitet af infektion (MOI) 0, 1 eller 10 (Sanini et al., 2018). Celler indsamlet på fire forskellige tidspunkter (4, 12, 24 og 48 timer efter infektion) blev derefter sorteret til enkeltcelletranskriptomisk analyse med en tilpasset smart-sek2-protokol., 2018). DENV – datasættet omfatter 933 inficerede celler (MOI = 1 eller 10) og 303 kontroller (MOI = 0), mens datasættet består af 488 inficerede celler (MOI = 1) og 403 kontroller. Begge datasæt blev log10 transformeret, og gener, der ikke blev udtrykt i mere end 80% af prøverne, blev fjernet. Datasættene blev derefter opdelt efter virus og efter tidspunkt og brugt som input (“Ekspressionsfil” – felt) til
vores semitool-analyser genererede i gennemsnit seks moduler pr.tidspunkt i denv-infektion og mere end otte moduler pr. tidspunkt i DENV-infektion. Vi har valgt et modul pr. tidspunkt som repræsentant for vores resultater (figur 2a). Det er klart, at ekspressionsaktiviteten af repræsentative moduler ved 24 og 48 timer efter infektion øges i henhold til virusbelastningen (figur 2a). Vi udførte derefter vejberigelsesanalysen af de repræsentative moduler ved 24 timer efter infektion ved hjælp af internetcemitool-linket til Enrichr (figur 2b). Disse resultater bekræfter ikke kun det, der blev beskrevet i den oprindelige publikation (Janini et al., 2018), men giver også ny indsigt i fysiopatologien af dengue-og Jika-virusinfektioner.
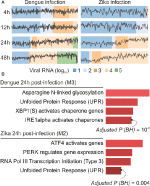
figur 2. en enkelt celle-RNA-sekv-data. (A) profil plot af co-udtrykte genmoduler. Vi valgte et repræsentativt modul for hvert tidspunkt post-dengue virus infektion (venstre) eller post-dengue virus infektion (højre). Den sorte linje repræsenterer medianekspressionsaktiviteten af gener fra modulerne på tværs af hver prøve. Farverne repræsenterer den forskellige mængde virus-RNA i cellen. (B) overrepræsentationsanalyse af udvalgte moduler ved 24 timer efter virusinfektion. Søjlediagrammerne blev tilpasset fra Enrichr-værktøjet, der er knyttet til enrichr-værktøjet. Søjlerne er proportional med-log10 justeret p-værdi (Benjamini-Hochberg) af de berigede veje i et modul.
Diskussion
selvom få lignende internetbaserede applikationer blev udviklet til at udføre co-ekspressionsgenanalyse., 2016; Desai et al., 2017), disse værktøjer giver ikke sammenlignelige resultater til
vi viste også, at vi er i stand til at analysere enkeltcellede RNA-sek-data hurtigere og effektivt. Vores resultater returnerede relevant information om de biologiske processer, der er involveret i dengue-og Jika-virusinfektion. Al denne analyse blev udført på en automatiseret og praktisk måde uden behov for brugeren at have dyb forståelse for den interne behandling af Gen-Co-ekspressionsdataanalyse.
Forfatterbidrag
LC, PR, BG-C og MA-P udførte analyserne. LC, GS-H og VM-C udviklede værktøjet. HN udtænkte værktøjet og overvågede arbejdet. Alle forfattere hjælper med at skrive papiret.
interessekonflikt Erklæring
forfatterne erklærer, at forskningen blev udført i mangel af kommercielle eller økonomiske forhold, der kunne fortolkes som en potentiel interessekonflikt.