Frontiers in Genetics
Introduction
細胞プロセスは、活性レベルを動的に調節しなければならない複数の相互作用分子によって駆動される(Kitano,2002)。 その結果、同じシグナル伝達および代謝経路に属する、または同様の機能を共有する遺伝子は、条件を越えて共発現される傾向がある(Wang et al., 2016). 共発現遺伝子モジュール解析は、その発現が高度に相関している遺伝子(すなわち、モジュール)のセットを含むネットワークを作成します。 このような分析は、感染性に関連する機能モジュールを明らかにするために適用された(Janova e t a l. ら、2 0 1 5)、炎症性(Beins e t a l. ら、2 0 1 6)、および神経学的(Voineagu e t a l. ら、2 0 1 1)疾患、ならびにいくつかのタイプの癌(Sharma e t a l., 2017).
Weighted gene co-expression network analysis(WGCNA)は、共発現遺伝子モジュールを同定するために広く使用されている方法です(Zhang and Horvath,2005)。 しかし、WGCNAを実行するためには、ユーザーはプログラミング環境に精通しているだけでなく、手動でパラメータを選択する必要があります。 これらの特徴はトランスクリプトームのデータセットからの遺伝子モジュールを識別するためにRの不十分な知識の研究者を防ぐ。
は、CEMiToolというバイオ伝導体Rパッケージに基づいています(Russo et al. これにより、バイオインフォマティクスのバックグラウンドを持たない科学者が包括的な共発現ネットワーク解析を実行できるようになりました。
材料と方法
webCEMiToolのwebインターフェイスは、ユーザーが特定のプログラムやインターネットブラウザをインストールすることなく、包括的な分析を迅速に生成できるように開発されました。 モジュラー分析を実行するための唯一の要件は、異なる生物学的条件下(本明細書では「クラス」として定義される)の試料中のすべての遺伝子の発現レベ サンプルの定義された範囲数はありませんが、私たちの以前の研究では、データセットあたり最低15サンプルが示唆されています(Russo et al., 2018). それは主にトランスクリプトームデータ(すなわち、RNA-seqまたはマイクロアレイ)のために設計されたが、それはまた、潜在的にタンパク質、サイトカイン、さらには代謝産物のモジュールを識別するために使用することができます。 その後、webCEMiToolは自動的に入力遺伝子を選択し、共発現モジュールを識別します。 各モジュールには、その発現が同様のパターンに従う遺伝子のセットが含まれています。
我々は、webCEMiTool内で、サンプルの各クラスの遺伝子モジュールの活性を評価する機能を実装しました。 このためには、各サンプルのクラスに通知するサンプル注釈タブ区切りのテキストファイルを提供するだけで済みます。 モジュール内の個々の遺伝子の中央値を示す”プロファイルプロット”は、ツールの”結果”セクションに表示されます(図1A)。
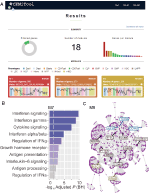
図1. webCEMiToolの概要. (A)webCEMiTool結果の概要–ドーナツチャートは、教師なしフィルタによって選択された遺伝子の割合を表します。 フロントページには、得られたモジュールの数と、各モジュールの遺伝子の数を示す棒グラフも表示されます。 モジュールプロファイルプロットは、各サンプルにわたるモジュールからの遺伝子の発現活性の中央値を示しています。 色は、さまざまなサンプルクラスを表します。 (B)過剰発現分析−これは、モジュール内の濃縮経路(ユーザ入力によって定義される経路)の−log1 0調整されたp値(Benjamini−Hochberg)を示す。gmtファイル)。 (C)モジュールの遺伝子ネットワーク–最も接続されている遺伝子(ハブ)は、最初にモジュールに存在していたか(青)、またはユーザーが入力した相互作用ファイル(赤)、ま
機能解析を可能にするために、遺伝子モジュールが特定のシグナル伝達経路または代謝経路に関連しているかどうかを確認することもできます(図1B)。 これらの経路は、KEGG、Reactome、MySigDBなどのデータベースから簡単に抽出できます。 最後に、ユーザーはinteractomeデータ(すなわち、タンパク質-タンパク質相互作用、転写因子とその転写遺伝子、あるいはmirnaとその標的遺伝子)と結果を統合することがで この機能により、ユーザーはモジュールの重要な規制当局を特定することができ(図1C)、実験的検証や薬物の潜在的な標的のための貴重な洞察を提供します。 オプションのファイルを取得する方法の詳細については、ウェブサイトの”チュートリアル”ページを参照してくださ2
我々の方法が堅牢であることを実証するために、我々はCEMiTool Rパッケージバージョンを使用して、1,000以上の公に利用可能なRNA-seqおよびマイクロアレイデータセッ, 2018). WebCEMiToolとパッケージには異なる視覚化機能があり、異なるプラットフォームに基づいていますが、コアの共表現機能は本質的に同じです。 ここで説明しているオンラインツールは、非プログラミング研究者のための遺伝子モジュラー解析への容易なアクセスを可能にするために構築されていますが、Rライブラリバージョンは、Rプログラミング言語のより大きな知識を持つユーザーを対象としています。 さらに、結果ダッシュボードは、解釈を容易にする対話型のグラフで構成されています。 さらに、バイオインフォマティクスwebサービスの上昇エコシステムを利用して、私たちのツールはEnrichrプラットフォームとのインターフェイスを確立します(Chen et al.,2013),私たちのユーザーのためのより豊かな経験を可能にします.
結果
私たちは、webCEMiToolが単一セルレベルで発現データを分析するために適用できることを実証しました。 公的に入手可能なviscRNA-Seqデータ(ウイルスを含む単一細胞RNA-Seq)は、NCBI GEOデータベース(受託番号GSE110496)から入手し、分析のための入力として使用した。 データは、感染の多重度(MOI)0、1、または1 0を使用して、デング熱ウイルス(DENV)またはZikaウイルス(ZIKV)のいずれかに感染した個々のヒト肝細胞癌(Huh7)細胞のトランスクリプトームを指す(Zanini e t a l., 2018). 次いで、4つの異なる時点(感染後4、1 2、2 4および4 8時間)に収集された細胞を、適応されたSmart−seq2プロトコル(Zanini e t a l.,2 0 0 2)を用いて、単一細胞転写分析のために分取した(Zanini e t a l.,2 0 0 2)。, 2018). DENVデータセットは、9 3 3個の感染細胞(MOI=1又は1 0)及び3 0 3個の対照(MOI=0)を含み、ZIKVデータセットは、4 8 8個の感染細胞(MOI=1)及び4 0 3個の対照から構成される。 分析をwebCEMiToolプラットフォームに提出する前に、両方のデータセットをlog10形質転換し、サンプルの80%以上で発現していない遺伝子を除去した。 次に、データセットをウイルスおよび時点で分割し、webCEMiToolへの入力(「式ファイル」フィールド)として使用しました。 遺伝子発現データに加えて、我々はまた、サンプル表現型をwebCEMiToolに提供しました(すなわち、ウイルス負荷)およびReactome遺伝子セット。
私たちのwebCEMiTool分析では、DENV感染では平均6つのモジュールが発生し、ZIKV感染では平均8つ以上のモジュールが発生しました。 私たちの調査結果の代表として、時点ごとに一つのモジュールを選択しました(図2A)。 感染後24時間および48時間で、代表的なモジュールの発現活性はウイルス量に応じて増加することは明らかである(図2A)。 次に、感染後24時間で、EnrichrのためのwebCEMiToolリンクを使用して、代表的なモジュールの経路濃縮分析を行った(図2B)。 これらの知見は、元の出版物に記載されたものを裏付けるだけでなく(Zanini e t a l. しかし、デング熱およびジカウイルス感染の生理学的病理学についての新しい洞察も提供する。
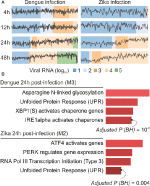
図2. webCEMiToolは、単一細胞RNA-seqデータに適用されます。 (A)共発現遺伝子モジュールのプロファイルプロット。 デングウイルス感染後(左)またはジカウイルス感染後(右)の各時点について、一つの代表的なモジュールを選択しました。 黒い線は、各サンプルにわたるモジュールからの遺伝子の発現活性の中央値を表す。 色は、細胞内のウイルスRNAの異なる量を表します。 (B)ウイルス感染後24時間における選択されたモジュールの過剰表現分析。 棒グラフは、webCEMiToolにリンクされたEnrichr webtoolから適応されました。 バーは、モジュール内の濃縮された経路の−log10調整されたp値(Benjamini-Hochberg)に比例する。
議論
共発現遺伝子解析を行うための類似のウェブベースのアプリケーションはほとんど開発されていないが(Tzfadia et al. ら、2 0 1 6;Desai e t a l. これらのツールは、webCEMiToolと同等の結果を提供しません。 そのような応用の1つは、Genet(Desai e t a l., 2017). このwebtoolは、遺伝子共発現解析を容易にするために設計され、濃縮分析と遺伝子間ネットワークを提供します。 しかし、これらの分析は3つの生物(R.capsulatus、m.tuberculosis、およびO.sativa)に対してのみ実行されます。 別の例は、Coexpnetviz(Tzfadia e t a l.,2016),遺伝子ネットワークの可視化と構築のために設計されたwebtool. GeNETと同様に、CoExpNetVizは、主に植物トランスクリプトームのために設計されていると述べられているように、生物に関して幾分限定されている。 WebCEMiToolは、任意の生物のための共発現解析を提供することを目指しています。 さらに、CoExpNetVizはwebベースのアプリケーションとして表示されますが、その結果はREADMEを含む圧縮フォルダとしてユーザーに返されます。cytoscapeアプリ上でその結果を視覚化する方法についての指示とtxtファイル。 ユーザーは、ツールによって提供されるいくつかの異なる出力ファイルをCytoscapeに手動で挿入する必要があります。 これらの追加の手順は、Cytoscapeに慣れていないユーザーにプロセスエラーが発生しやすく、おそらく困難になる可能性があります。 WebCEMiToolは、はるかに便利なブラウザ表示の結果を提供しています。
また、webCEMitoolが単細胞RNA-seqデータをより迅速かつ効率的に分析できることを示しました。 我々の結果は、デング熱およびジカウイルス感染に関与する生物学的プロセスに関する関連情報を返した。 すべての解析は、ユーザーが遺伝子共発現データ解析の内部処理について深い理解を持っている必要はなく、自動化された実用的な方法で行われました。
著者の貢献
LC、PR、BG-C、MA-Pが分析を行った。 LC、GS-H、VM-Cはwebtoolを開発しました。 HNはツールを考案し、作業を監督しました。 すべての著者は論文の執筆に役立ちます。
利益相反声明
著者らは、この研究は利益相反の可能性と解釈される可能性のある商業的または財政的関係がない場合に行われたと宣言している。