Frontiers in Genetics
Introduction
cellulaire processen worden gedreven door meerdere interagerende moleculen waarvan het activiteitsniveau dynamisch moet worden gereguleerd (Kitano, 2002). Dientengevolge, zullen de genen die tot dezelfde signalerende en metabolische weg behoren of gelijkaardige functies delen neigen om over voorwaarden (Wang et al., 2016). De analyse van de co-expressiegenmodule leidt tot netwerken die reeksen genen (d.w.z., modules) omvatten waarvan de uitdrukking hoogst gecorreleerd is. Dergelijke analyse werd toegepast om functionele modules met betrekking tot infectieuze onthullen (Janova et al., 2015), inflammatoire (Beins et al., 2016), en neurologisch (Voineagu et al., 2011) ziekten, evenals verschillende soorten kanker (Sharma et al., 2017).Weighted gen co-expression network analysis (Wgcna) is een veel gebruikte methode om co-expression genmodules te identificeren (Zhang and Horvath, 2005). Om WGCNA uit te voeren, moeten gebruikers echter vertrouwd zijn met programmeeromgevingen en handmatig parameters selecteren. Deze eigenschappen verhinderen onderzoekers met onvoldoende kennis van R om genmodules van transcriptome gegevensreeksen te identificeren.
gebaseerd op ons Bioconductor R pakket genaamd CEMiTool (Russo et al., 2018), ontwikkelden we een gebruiksvriendelijke web-based applicatie die wetenschappers zonder achtergrond in bioinformatica in staat stelt om uitgebreide Co-expressienetwerk analyse uit te voeren.
materialen en methoden
de webinterface van webCEMiTool is ontwikkeld om gebruikers in staat te stellen snel uitgebreide analyses te genereren zonder dat er een specifiek programma of internetbrowser hoeft te worden geïnstalleerd. De enige vereiste voor het uitvoeren van de modulaire analyse is een gegevensverzameling met de expressieniveaus van alle genen in monsters onder verschillende biologische omstandigheden (hierna gedefinieerd als “klassen”). Er is geen gedefinieerd bereik aantal monsters, maar onze vorige studie suggereert een minimum van 15 monsters per dataset (Russo et al., 2018). Hoewel het hoofdzakelijk voor transcriptome gegevens (d.w.z., RNA-seq of microarrays) werd ontworpen, kan het potentieel ook voor het identificeren van modules van proteã nen, cytokines, en zelfs metabolites worden gebruikt. webCEMiTool selecteert dan automatisch de invoergenen en identificeert de co-expressiemodules. Elke module bevat een reeks genen waarvan de uitdrukking een gelijkaardig patroon volgt.
binnen webCEMiTool hebben we een functie geïmplementeerd die de activiteit van genmodules op elke klasse monsters beoordeelt. Hiervoor hoeven de gebruikers alleen een door het tabblad voorbeeldannotatie gescheiden tekstbestand te verstrekken dat de klasse van elk voorbeeldbestand informeert. Een ” profielplot “die het mediane niveau van individuele genen binnen de module toont, wordt dan weergegeven in de sectie” resultaten ” van het hulpmiddel (figuur 1A).
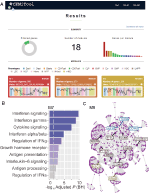
figuur 1. webCEMiTool overzicht. (A) webcemitool results summary – de donut grafiek geeft het aandeel van geselecteerde genen door de zonder toezicht filter. De voorpagina toont ook het aantal modules verkregen, evenals een staafdiagram beeltenis van het aantal genen in elke module. De plots van het moduleprofiel illustreren de mediane uitdrukkingsactiviteit van genen van de modules over elke steekproef. De kleuren vertegenwoordigen de verschillende monsterklassen. (B) Overrepresentatie analyse – Dit toont de −log10 aangepaste p-waarde (Benjamini-Hochberg) van de verrijkte paden in een module (paden gedefinieerd door de gebruiker ingevoerd .gmt-bestand). (C) gen netwerk van een module – de top meest verbonden genen (hubs) worden geëtiketteerd en gekleurd op basis van de vraag of ze oorspronkelijk aanwezig waren in de module (blauw), of ingevoegd uit een gebruiker ingevoerde interactie bestand (rood), of beide (groen).
om functionele analyse toe te laten, kunnen de gebruikers ook controleren of de genmodules met specifieke signalerende of metabolische wegen worden geassocieerd (figuur 1B). Deze wegen kunnen gemakkelijk uit databases, zoals KEGG, Reactome, en MySigDB worden gehaald. Tot slot, kunnen de gebruikers de resultaten met interactome gegevens (d.w.z., eiwit-eiwitinteractie, transcriptiefactoren en hun getranscribeerde genen, of zelfs miRNAs en hun doelgenen integreren). Deze eigenschap stelt gebruikers in staat om kritieke regelgevers van modules (figuur 1C) te identificeren, die waardevolle inzichten voor experimentele validatie of potentiële doelstellingen voor drugs verstrekken. Meer informatie over het verkrijgen van de optionele bestanden vindt u op de “Tutorial” pagina van de website.2
om aan te tonen dat onze methode robuust is, hebben we een ongekende grootschalige modulaire analyse uitgevoerd met meer dan 1.000 publiek beschikbare RNA-seq en microarray data sets en nieuwe RNA-seq data van patiënten die geïnfecteerd zijn met Leishmania met behulp van de cemitool R package version (Russo et al., 2018). Hoewel webCEMiTool en het pakket hebben verschillende visualisatie functies en zijn gebaseerd op verschillende platforms, de kern co-expressie functionaliteit is in wezen hetzelfde. De online tool die we hier beschrijven is gebouwd om gemakkelijke toegang tot Gen modulaire analyses voor niet-programmerende onderzoekers mogelijk te maken, terwijl de R-bibliotheekversie is gericht op Gebruikers met een grotere kennis van de R-programmeertaal. Bovendien, de resultaten dashboard is samengesteld uit interactieve grafieken die interpretatie te vergemakkelijken. Bovendien, gebruik te maken van de stijgende ecosysteem van Bioinformatica web services, Onze tool stelt een interface met de Enrichr platform (Chen et al., 2013), waardoor een rijkere ervaring voor onze gebruikers.
resultaten
we hebben aangetoond dat webCEMiTool kan worden toegepast om expressiegegevens op eencellig niveau te analyseren. Publiek beschikbare viscRNA-Seq-gegevens (virus-met inbegrip van single cell RNA-Seq) werden verkregen uit de NCBI GEO-database (toetredingsnummer GSE110496) en gebruikt als input voor de analyse. De gegevens hebben betrekking op het transcriptoom van individuele humane hepatoom (Huh7) cellen, die besmet waren met ofwel dengue virus (DENV) of Zika virus (ZIKV), met behulp van multiplicity of infection (MOI) 0, 1, of 10 (Zanini et al., 2018). Cellen verzameld op vier verschillende tijdstippen (4, 12, 24 en 48 uur na infectie) werden vervolgens gesorteerd voor eencellige transcriptomische analyse met een aangepast Smart-seq2 protocol (Zanini et al., 2018). De denv-gegevensverzameling bestaat uit 933 geïnfecteerde cellen (MOI = 1 of 10) en 303 controles (MOI = 0), terwijl de ZIKV-gegevensverzameling bestaat uit 488 geïnfecteerde cellen (MOI = 1) en 403 controles. Voordat de analyse aan het webCEMiTool platform werd voorgelegd, werden beide datasets log10 getransformeerd en werden genen verwijderd die niet in meer dan 80% van de steekproeven werden uitgedrukt. De datasets werden vervolgens gesplitst door virus en door tijdstip en gebruikt als invoer (“Expression file” veld) naar webCEMiTool. Naast de gegevens van de genuitdrukking, verstrekten wij ook aan webCEMiTool de steekproeffenotypes (d.w.z., virale ladingen) en Reactome gensets.
onze webcemitool-analyses genereerden gemiddeld zes modules per tijdstip in DENV-infectie en meer dan acht modules per tijdstip in ZIKV-infectie. We hebben één module per tijdstip geselecteerd als vertegenwoordiger van onze bevindingen (figuur 2A). Het is duidelijk dat bij 24 en 48 uur na de infectie de expressieactiviteit van representatieve modules toeneemt naargelang van de virale belasting (figuur 2A). Vervolgens voerden we de pathway enrichment analyse uit van de representatieve modules op 24 uur na infectie met behulp van de webCEMiTool link voor Enrichr (figuur 2B). Deze bevindingen bevestigen niet alleen wat in de oorspronkelijke publicatie werd beschreven (Zanini et al., 2018) maar bieden ook nieuwe inzichten over de fysiopathologie van dengue-en Zika-virusinfecties.
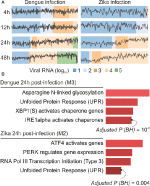
Figuur 2. webCEMiTool toegepast op single-cell RNA-seq gegevens. (A) Profielplot van co-tot expressie gebrachte genmodules. We selecteerden een representatieve module voor elk tijdstip post-dengue virus infectie (links) of post-Zika virus infectie (rechts). De zwarte lijn vertegenwoordigt de mediane uitdrukkingsactiviteit van genen van de modules over elke steekproef. De kleuren vertegenwoordigen de verschillende hoeveelheid virusrna binnen de cel. B) analyse van de oververtegenwoordiging van geselecteerde modules op 24 uur na de virusinfectie. De staafgrafieken zijn aangepast van de Enrichr webtool gekoppeld aan webCEMiTool. De balken zijn evenredig met de-log10 aangepaste p-waarde (Benjamini-Hochberg) van de verrijkte paden in een module.
discussie
hoewel er weinig vergelijkbare webgebaseerde toepassingen werden ontwikkeld om co-expressiegenanalyse uit te voeren (Tzfadia et al., 2016; Desai et al., 2017), bieden deze tools geen vergelijkbare resultaten als webCEMiTool. Een dergelijke toepassing is GeNET (Desai et al., 2017). Deze webtool werd ontworpen om gen co-expressie analyses te vergemakkelijken en biedt verrijking analyse en gen-aan-gen netwerken. Deze analyses worden echter slechts uitgevoerd voor drie organismen (R. capsulatus, M. tuberculosis en O. sativa). Een ander voorbeeld is CoExpNetViz (Tzfadia et al., 2016), een webtool ontworpen voor de visualisatie en bouw van gennetwerken. Net als bij GeNET is CoExpNetViz enigszins beperkt met betrekking tot de organismen, aangezien wordt beweerd dat het voornamelijk is ontworpen voor plantentranscriptomen. Het webCEMiTool heeft als doel om co-expressieanalyses te leveren voor elk organisme. Bovendien, hoewel CoExpNetViz wordt gepresenteerd als een web-based applicatie, de resultaten worden geretourneerd aan gebruikers als een gecomprimeerde map met een README.txt-bestand met instructies voor het visualiseren van hun resultaten op de Cytoscape app. De gebruikers moeten dan handmatig invoegen in Cytoscape de verschillende output bestanden die door de tool. Deze extra stappen kunnen ook het proces foutgevoelig en mogelijk ontmoedigend voor gebruikers onbekend met Cytoscape. De webCEMiTool biedt veel handiger browser-weergegeven resultaten.
we toonden ook aan dat webCEMitool in staat is om single-cell RNA-seq data sneller en efficiënt te analyseren. Onze resultaten retourneerden relevante informatie over de biologische processen die betrokken zijn bij dengue-en Zika-virusinfectie. Al deze analyse werd uitgevoerd op een geautomatiseerde en praktische manier, zonder behoefte aan de gebruiker om diep begrip over de interne verwerking van de gegevensanalyse van de Genco-uitdrukking te hebben.
bijdragen van auteurs
LC, PR, BG-C en MA-P hebben de analyses uitgevoerd. LC, GS-H en VM-C ontwikkelden de webtool. HN bedacht het gereedschap en begeleidde het werk. Alle auteurs helpen bij het schrijven van het papier.
belangenverstrengeling verklaring
de auteurs verklaren dat het onderzoek werd uitgevoerd zonder enige commerciële of financiële relatie die als een potentieel belangenconflict kon worden opgevat.