Fronteras en Genética
Introducción
Los procesos celulares son impulsados por múltiples moléculas que interactúan cuyo nivel de actividad debe regularse dinámicamente (Kitano, 2002). Como resultado, los genes que pertenecen a la misma vía de señalización y metabólica o que comparten funciones similares tenderán a ser coexpresados en todas las condiciones (Wang et al., 2016). El análisis de módulos genéticos de coexpresión crea redes que comprenden conjuntos de genes (es decir, módulos) cuya expresión está altamente correlacionada. Dicho análisis se aplicó para revelar módulos funcionales relacionados con infecciones (Janova et al., 2015), inflamatorio (Beins et al., 2016) y neurológica (Voineagu et al., 2011), así como varios tipos de cáncer (Sharma et al., 2017).
El análisis de red de coexpresión de genes ponderada (WGCNA) es un método ampliamente utilizado para identificar módulos de genes coexpresión (Zhang y Horvath, 2005). Sin embargo, para ejecutar WGCNA, los usuarios deben estar familiarizados con los entornos de programación, así como seleccionar parámetros manualmente. Estas características impiden que los investigadores con un conocimiento insuficiente de R identifiquen módulos genéticos a partir de conjuntos de datos de transcriptomas.
Basado en nuestro paquete Bioconductor R llamado CEMiTool (Russo et al., 2018), desarrollamos una aplicación web fácil de usar que permite a los científicos sin experiencia en bioinformática realizar un análisis de red de coexpresión integral.
Materiales y métodos
La interfaz web de webCEMiTool fue desarrollada para permitir a los usuarios generar análisis exhaustivos rápidamente sin necesidad de instalar ningún programa específico o navegador de Internet. El único requisito para ejecutar el análisis modular es un conjunto de datos que contenga los niveles de expresión de todos los genes en muestras bajo diferentes condiciones biológicas (en este documento se definen como “clases”). No hay un número de muestras de rango definido, pero nuestro estudio previo sugiere un mínimo de 15 muestras por conjunto de datos (Russo et al., 2018). Aunque fue diseñado principalmente para datos de transcriptomas (es decir, ARN-seq o microarrays), también puede usarse potencialmente para identificar módulos de proteínas, citoquinas e incluso metabolitos. webCEMiTool seleccionará automáticamente los genes de entrada e identificará los módulos de coexpresión. Cada módulo contiene un conjunto de genes cuya expresión sigue un patrón similar.
Implementamos, dentro de webCEMiTool, una característica que evalúa la actividad de los módulos genéticos en cada clase de muestras. Para ello, los usuarios solo tienen que proporcionar un archivo de texto delimitado por tabulaciones de anotación de muestra que informa a la clase de cada muestra. Una “gráfica de perfil” que muestra el nivel medio de genes individuales dentro del módulo se muestra en la sección “Resultados” de la herramienta (Figura 1A).
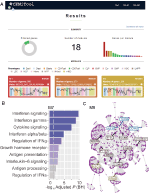
Figura 1. Visión general de webCEMiTool. (A) Resumen de resultados de webCEMiTool – La tabla de donas representa la proporción de genes seleccionados por el filtro no supervisado. La portada también muestra el número de módulos obtenidos, así como un gráfico de barras que representa el número de genes en cada módulo. Las gráficas de perfil de módulo ilustran la actividad de expresión media de los genes de los módulos en cada muestra. Los colores representan las diferentes clases de muestra. B) Análisis de sobrerrepresentación: representa el valor de p ajustado log10 (Benjamini-Hochberg) de las vías enriquecidas en un módulo (vías definidas por el usuario ingresado).archivo gmt). (C) Red de genes de un módulo: Los genes más conectados (hubs) se etiquetan y colorean en función de si estaban presentes originalmente en el módulo (azul), o insertados desde un archivo de interacción introducido por el usuario (rojo), o ambos (verde).
Para permitir el análisis funcional, los usuarios también pueden verificar si los módulos genéticos están asociados con vías metabólicas o de señalización específicas (Figura 1B). Estas vías se pueden extraer fácilmente de bases de datos, como KEGG, Reactome y MySigDB. Finalmente, los usuarios pueden integrar los resultados con los datos del interactoma (es decir, interacciones proteína-proteína, factores de transcripción y sus genes transcritos, o incluso miRNAs y sus genes diana). Esta característica permite a los usuarios identificar reguladores críticos de módulos (Figura 1C), lo que proporciona información valiosa para la validación experimental o los objetivos potenciales de los medicamentos. Puede encontrar más detalles sobre cómo obtener los archivos opcionales en la página “Tutorial” del sitio web.2
Para demostrar que nuestro método es robusto, realizamos un análisis modular a gran escala sin precedentes con más de 1.000 conjuntos de datos de ARN-seq y microarrays disponibles públicamente y nuevos datos de ARN-seq de pacientes infectados con Leishmania utilizando la versión del paquete CEMiTool R (Russo et al., 2018). Aunque webCEMiTool y el paquete tienen características de visualización distintas y se basan en plataformas diferentes, la funcionalidad principal de coexpresión es esencialmente la misma. La herramienta en línea que describimos aquí está diseñada para permitir un fácil acceso a los análisis modulares de genes para investigadores que no son programadores, mientras que la versión de la biblioteca R está dirigida a usuarios con un mayor conocimiento del lenguaje de programación R. Además, el panel de resultados se compone de gráficos interactivos que facilitan la interpretación. Además, aprovechando el creciente ecosistema de servicios web bioinformáticos, nuestra herramienta establece una interfaz con la plataforma Enrichr (Chen et al., 2013), lo que permite una experiencia más rica para nuestros usuarios.
Resultados
Demostramos que webCEMiTool se puede aplicar para analizar datos de expresión a nivel de celda única. Los datos disponibles públicamente de viscRNA-Seq (virus, incluido el ARN unicelular-Seq) se obtuvieron de la base de datos NCBI GEO (número de acceso GSE110496) y se utilizaron como datos de entrada para el análisis. Los datos se refieren al transcriptoma de células de hepatoma humano individual (Huh7), que estaban infectadas con el virus del dengue (DENV) o el virus del Zika (ZIKV), utilizando multiplicidad de infecciones (MOI) 0, 1 o 10 (Zanini et al., 2018). Las células recolectadas en cuatro puntos de tiempo diferentes (4, 12, 24 y 48 h después de la infección) se clasificaron para el análisis transcriptómico unicelular con un protocolo Smart-seq2 adaptado (Zanini et al., 2018). El conjunto de datos DENV comprende 933 células infectadas ( MOI = 1 o 10) y 303 controles (MOI = 0), mientras que el conjunto de datos ZIKV se compone de 488 células infectadas (MOI = 1) y 403 controles. Antes de enviar el análisis a la plataforma webCEMiTool, ambos conjuntos de datos se transformaron log10 y se eliminaron los genes que no se expresaban en más del 80% de las muestras. Los conjuntos de datos se dividieron por virus y por punto de tiempo y se utilizaron como entrada (campo”Archivo de expresión”) en webCEMiTool. Además de los datos de expresión génica, también proporcionamos a webCEMiTool los fenotipos de muestra (p. ej., la carga viral) y Reactome conjuntos de genes.
Nuestros análisis de webCEMiTool generaron un promedio de seis módulos por punto de tiempo en la infección por DENV y más de ocho módulos por punto de tiempo en la infección por ZIKV. Hemos seleccionado un módulo por punto de tiempo como representante de nuestros hallazgos (Figura 2A). Está claro que a las 24 y 48 h post-infección, la actividad de expresión de los módulos representativos aumenta de acuerdo con la carga viral (Figura 2A). A continuación, realizamos el análisis de enriquecimiento de vías de los módulos representativos a las 24 h post-infección utilizando el enlace webCEMiTool para Enrichr (Figura 2B). Estos hallazgos no solo corroboran lo descrito en la publicación original (Zanini et al., 2018), pero también proporcionan nuevos conocimientos sobre la fisiopatología de las infecciones por el dengue y el virus del Zika.
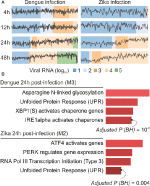
Figura 2. webCEMiTool aplicado a datos de ARN-seq unicelulares. A) Diagrama de perfil de módulos de genes coexpresados. Seleccionamos un módulo representativo para cada punto de tiempo posterior a la infección por el virus del dengue (izquierda) o posterior a la infección por el virus del Zika (derecha). La línea negra representa la actividad de expresión media de los genes de los módulos en cada muestra. Los colores representan la diferente cantidad de ARN del virus dentro de la célula. B) Análisis de la sobrerrepresentación de módulos seleccionados a las 24 horas después de la infección por el virus. Los gráficos de barras se adaptaron de la herramienta web Enrichr vinculada a webCEMiTool. Las barras son proporcionales al valor de p ajustado-log10 (Benjamini-Hochberg) de las vías enriquecidas en un módulo.
Discusión
Aunque se desarrollaron pocas aplicaciones similares basadas en la web para realizar análisis de genes de coexpresión (Tzfadia et al., 2016; Desai et al., 2017), estas herramientas no proporcionan resultados comparables a webCEMiTool. Una de esas aplicaciones es GeNET (Desai et al., 2017). Esta herramienta web fue diseñada para facilitar los análisis de coexpresión de genes y proporciona análisis de enriquecimiento y redes gene-to-gen. Sin embargo, solo realiza estos análisis para tres organismos (R. capsulatus, M. tuberculosis y O. sativa). Otro ejemplo es CoExpNetViz (Tzfadia et al., 2016), una herramienta web diseñada para la visualización y construcción de redes genéticas. Al igual que GeNET, CoExpNetViz es algo limitado con respecto a los organismos, ya que se afirma que está diseñado principalmente para transcriptomas de plantas. El webCEMiTool tiene como objetivo proporcionar análisis de coexpresión para cualquier organismo. Además, aunque CoExpNetViz se presenta como una aplicación basada en la web, sus resultados se devuelven a los usuarios como una carpeta comprimida que contiene un README.archivo txt con instrucciones sobre cómo visualizar sus resultados en la aplicación Cytoscape. Los usuarios tienen que insertar manualmente en Cytoscape los diferentes archivos de salida proporcionados por la herramienta. Estos pasos adicionales también pueden hacer que el proceso sea propenso a errores y posiblemente desalentador para los usuarios que no estén familiarizados con Cytoscape. webCEMiTool ofrece resultados mucho más convenientes que se muestran en el navegador.
También mostramos que webCEMitool es capaz de analizar datos de ARN-seq de una sola célula de forma más rápida y eficiente. Nuestros resultados arrojaron información relevante sobre los procesos biológicos involucrados con la infección por el dengue y el virus del Zika. Todos estos análisis se realizaron de manera automatizada y práctica, sin necesidad de que el usuario tuviera un conocimiento profundo del procesamiento interno del análisis de datos de coexpresión de genes.
Contribuciones de autores
LC, PR, BG-C y MA-P realizaron los análisis. LC, GS-H y VM-C desarrollaron la herramienta web. HN concibió la herramienta y supervisó el trabajo. Todos los autores ayudan en la redacción del artículo.
Declaración de Conflicto de Intereses
Los autores declaran que la investigación se realizó en ausencia de relaciones comerciales o financieras que pudieran interpretarse como un conflicto de intereses potencial.