Frontiers in Genetics
Introduction
Les processus cellulaires sont pilotés par de multiples molécules en interaction dont le niveau d’activité doit être régulé dynamiquement (Kitano, 2002). Par conséquent, les gènes appartenant à la même voie de signalisation et métabolique ou partageant des fonctions similaires auront tendance à être co-exprimés dans toutes les conditions (Wang et al., 2016). L’analyse par module de gènes de co-expression crée des réseaux comprenant des ensembles de gènes (c’est-à-dire des modules) dont l’expression est fortement corrélée. Une telle analyse a été appliquée pour révéler des modules fonctionnels liés aux maladies infectieuses (Janova et al., 2015), inflammatoire (Beins et al., 2016), et neurologiques (Voineagu et al., 2011) maladies, ainsi que plusieurs types de cancer (Sharma et al., 2017).
L’analyse pondérée du réseau de co-expression des gènes (WGCNA) est une méthode largement utilisée pour identifier les modules de gènes co-exprimés (Zhang et Horvath, 2005). Cependant, pour exécuter WGCNA, les utilisateurs doivent se familiariser avec les environnements de programmation et sélectionner manuellement les paramètres. Ces caractéristiques empêchent les chercheurs ayant une connaissance insuffisante de R d’identifier des modules de gènes à partir d’ensembles de données de transcriptome.
Basé sur notre paquet Bioconducteur R nommé CEMiTool (Russo et al., 2018), nous avons développé une application Web conviviale qui permet aux scientifiques sans expérience en bioinformatique d’effectuer une analyse complète du réseau de co-expression.
Matériaux et méthodes
L’interface web de webCEMiTool a été développée pour permettre aux utilisateurs de générer rapidement des analyses complètes sans avoir besoin d’installer un programme ou un navigateur Internet spécifique. La seule exigence pour exécuter l’analyse modulaire est un ensemble de données contenant les niveaux d’expression de tous les gènes dans des échantillons dans différentes conditions biologiques (ici définies comme des “classes”). Il n’y a pas de nombre défini d’échantillons, mais notre étude précédente suggère un minimum de 15 échantillons par ensemble de données (Russo et al., 2018). Bien qu’il ait été principalement conçu pour les données du transcriptome (c.-à-d. ARN-seq ou microarrays), il peut également être utilisé pour identifier des modules de protéines, de cytokines et même de métabolites. webCEMiTool sélectionnera alors automatiquement les gènes d’entrée et identifiera les modules de co-expression. Chaque module contient un ensemble de gènes dont l’expression suit un schéma similaire.
Nous avons implémenté, au sein de webCEMiTool, une fonctionnalité qui évalue l’activité des modules de gènes sur chaque classe d’échantillons. Pour cela, les utilisateurs n’ont qu’à fournir un exemple de fichier texte délimité par des onglets d’annotation qui informe la classe de chaque échantillon. Un “tracé de profil” montrant le niveau médian de gènes individuels au sein du module est ensuite affiché dans la section “Résultats” de l’outil (Figure 1A).
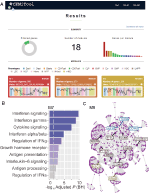
Figure 1. Présentation de l’outil webCEMiTool. (A) Résumé des résultats de webCEMiTool – Le diagramme en beignet représente la proportion de gènes sélectionnés par le filtre non supervisé. La première page affiche également le nombre de modules obtenus, ainsi qu’un graphique à barres représentant le nombre de gènes dans chaque module. Les diagrammes de profil de module illustrent l’activité d’expression médiane des gènes des modules dans chaque échantillon. Les couleurs représentent les différentes classes d’échantillons. (B) Analyse de la surreprésentation – Cela représente la valeur p ajustée -log10 (Benjamini-Hochberg) des voies enrichies dans un module (voies définies par l’utilisateur entrées.fichier gmt). (C) Réseau de gènes d’un module – Les gènes les plus connectés (hubs) sont étiquetés et colorés selon qu’ils étaient initialement présents dans le module (bleu) ou insérés à partir d’un fichier d’interaction entré par l’utilisateur (rouge), ou les deux (vert).
Pour permettre l’analyse fonctionnelle, les utilisateurs peuvent également vérifier si les modules génétiques sont associés à des voies de signalisation ou métaboliques spécifiques (Figure 1B). Ces voies peuvent facilement être extraites de bases de données, telles que KEGG, Reactome et MySigDB. Enfin, les utilisateurs peuvent intégrer les résultats aux données de l’interactome (c’est-à-dire les interactions protéine-protéine, les facteurs de transcription et leurs gènes transcrits, ou même les MIARN et leurs gènes cibles). Cette fonctionnalité permet aux utilisateurs d’identifier les régulateurs critiques des modules (figure 1C), fournissant des informations précieuses pour la validation expérimentale ou des cibles potentielles pour les médicaments. Des détails supplémentaires sur la façon d’obtenir les fichiers optionnels peuvent être trouvés dans la page “Tutoriel” du site Web.2
Pour démontrer que notre méthode est robuste, nous avons effectué une analyse modulaire à grande échelle sans précédent avec plus de 1 000 ensembles de données ARN-seq et microarray accessibles au public et de nouvelles données ARN-seq de patients infectés par la Leishmania en utilisant la version du package CEMiTool R (Russo et al., 2018). Bien que webCEMiTool et le package aient des fonctionnalités de visualisation distinctes et soient basés sur des plates-formes différentes, la fonctionnalité de co-expression de base est essentiellement la même. L’outil en ligne que nous décrivons ici est conçu pour permettre un accès facile aux analyses modulaires de gènes pour les chercheurs non programmeurs, tandis que la version de la bibliothèque R est destinée aux utilisateurs ayant une plus grande connaissance du langage de programmation R. De plus, le tableau de bord des résultats est composé de graphiques interactifs qui facilitent l’interprétation. De plus, profitant de l’écosystème croissant des services web bioinformatiques, notre outil établit une interface avec la plateforme Enrichr (Chen et al., 2013), permettant une expérience plus riche pour nos utilisateurs.
Résultats
Nous avons démontré que webCEMiTool peut être appliqué pour analyser des données d’expression au niveau d’une seule cellule. Les données de viscRNA-Seq accessibles au public (virus – y compris l’ARN unicellulaire-Seq) ont été obtenues à partir de la base de données NCBI GEO (numéro d’accession GSE110496) et utilisées comme entrée pour l’analyse. Les données se réfèrent au transcriptome de cellules individuelles d’hépatome humain (Huh7), infectées par le virus de la dengue (DENV) ou le virus Zika (ZIKV), en utilisant multiplicité d’infection (MOI) 0, 1 ou 10 (Zanini et al., 2018). Les cellules collectées sur quatre temps différents (4, 12, 24 et 48 h après l’infection) ont ensuite été triées pour une analyse transcriptomique unicellulaire avec un protocole Smart-seq2 adapté (Zanini et al., 2018). L’ensemble de données DENV comprend 933 cellules infectées (MOI = 1 ou 10) et 303 témoins (MOI = 0), tandis que l’ensemble de données ZIKV est composé de 488 cellules infectées (MOI = 1) et de 403 témoins. Avant de soumettre l’analyse à la plateforme webCEMiTool, les deux ensembles de données ont été transformés en log10 et les gènes qui n’étaient pas exprimés dans plus de 80% des échantillons ont été retirés. Les ensembles de données ont ensuite été divisés par virus et par point temporel et utilisés comme entrée (champ “Fichier d’expression”) dans webCEMiTool. En plus des données d’expression génique, nous avons également fourni à webCEMiTool les phénotypes d’échantillons (i.e., charges virales) et des ensembles de gènes du réactome.
Nos analyses webCEMiTool ont généré en moyenne six modules par point temporel dans l’infection à DENV et plus de huit modules par point temporel dans l’infection à ZIKV. Nous avons sélectionné un module par point temporel comme représentant de nos résultats (figure 2A). Il est clair qu’à 24 et 48 h post-infection, l’activité d’expression des modules représentatifs augmente en fonction de la charge virale (Figure 2A). Nous avons ensuite effectué l’analyse d’enrichissement de la voie des modules représentatifs 24 h après l’infection en utilisant le lien webCEMiTool pour Enrichr (Figure 2B). Ces résultats corroborent non seulement ce qui a été décrit dans la publication originale (Zanini et al., 2018) mais fournissent également de nouvelles informations sur la physiopathologie des infections par la dengue et le virus Zika.
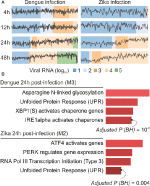
Figure 2. webCEMiTool appliqué aux données d’ARN-seq unicellulaires. (A) Tracé du profil des modules de gènes co-exprimés. Nous avons sélectionné un module représentatif pour chaque point temporel après l’infection par le virus de la dengue (à gauche) ou après l’infection par le virus Zika (à droite). La ligne noire représente l’activité d’expression médiane des gènes des modules de chaque échantillon. Les couleurs représentent la quantité différente d’ARN viral dans la cellule. (B) Analyse de la surreprésentation de certains modules 24 h après l’infection par le virus. Les graphiques à barres ont été adaptés du webtool Enrichr lié à webCEMiTool. Les barres sont proportionnelles à la valeur p ajustée -log10 (Benjamini-Hochberg) des voies enrichies dans un module.
Discussion
Bien que peu d’applications Web similaires aient été développées pour effectuer une analyse des gènes de co-expression (Tzfadia et al., 2016; Desai et coll., 2017), ces outils ne fournissent pas de résultats comparables à webCEMiTool. L’une de ces applications est GeNET (Desai et al., 2017). Ce webtool a été conçu pour faciliter les analyses de co-expression de gènes et fournit une analyse d’enrichissement et des réseaux de gènes à gènes. Cependant, il ne réalise ces analyses que pour trois organismes (R. capsulatus, M. tuberculosis et O. sativa). Un autre exemple est CoExpNetViz (Tzfadia et al., 2016), un outil web conçu pour la visualisation et la construction de réseaux de gènes. Semblable à GeNET, CoExpNetViz est quelque peu limité en ce qui concerne les organismes, car il est déclaré qu’il est principalement conçu pour les transcriptomes végétaux. Le webCEMiTool vise à fournir des analyses de co-expression pour tout organisme. De plus, bien que CoExpNetViz soit présenté comme une application Web, ses résultats sont renvoyés aux utilisateurs sous la forme d’un dossier compressé contenant un fichier README.fichier txt avec des instructions sur la façon de visualiser leurs résultats sur l’application Cytoscape. Les utilisateurs doivent ensuite insérer manuellement dans Cytoscape les différents fichiers de sortie fournis par l’outil. Ces étapes supplémentaires peuvent également rendre le processus sujet aux erreurs et peut-être intimidant pour les utilisateurs peu familiers avec Cytoscape. L’outil webCEMiTool offre des résultats affichés par navigateur beaucoup plus pratiques.
Nous avons également montré que webCEMitool est capable d’analyser les données d’ARN-seq unicellulaires plus rapidement et efficacement. Nos résultats ont fourni des informations pertinentes sur les processus biologiques impliqués dans l’infection par la dengue et le virus Zika. Toutes ces analyses ont été effectuées de manière automatisée et pratique, sans qu’il soit nécessaire pour l’utilisateur d’avoir une compréhension approfondie du traitement interne de l’analyse des données de co-expression des gènes.
Contributions des auteurs
LC, PR, BG-C et MA-P ont effectué les analyses. Le webtool a été développé par LC, GS-H et VM-C. HN a conçu l’outil et supervisé le travail. Tous les auteurs aident à la rédaction de l’article.
Déclaration de conflit d’intérêts
Les auteurs déclarent que la recherche a été menée en l’absence de relations commerciales ou financières pouvant être interprétées comme un conflit d’intérêts potentiel.