Frontiers in Genetics
Einleitung
Zelluläre Prozesse werden von mehreren interagierenden Molekülen gesteuert, deren Aktivitätsniveau dynamisch reguliert werden muss (Kitano, 2002). Infolgedessen neigen Gene, die zu demselben Signal- und Stoffwechselweg gehören oder ähnliche Funktionen teilen, dazu, unter verschiedenen Bedingungen coexprimiert zu werden (Wang et al., 2016). Die Koexpressionsgenmodulanalyse erzeugt Netzwerke, die Sätze von Genen (d. H. Modulen) umfassen, deren Expression stark korreliert ist. Eine solche Analyse wurde angewendet, um funktionelle Module im Zusammenhang mit infektiösen (Janova et al., 2015), entzündliche (Beins et al., 2016) und neurologisch (Voineagu et al., 2011) Krankheiten sowie verschiedene Krebsarten (Sharma et al., 2017).
Die gewichtete Gen-Co-Expressions-Netzwerkanalyse (WGCNA) ist eine weit verbreitete Methode zur Identifizierung von co-exprimierten Genmodulen (Zhang und Horvath, 2005). Um WGCNA ausführen zu können, müssen Benutzer jedoch mit Programmierumgebungen vertraut sein und Parameter manuell auswählen. Diese Merkmale verhindern, dass Forscher mit unzureichenden Kenntnissen von R Genmodule aus Transkriptomdatensätzen identifizieren können.
Basierend auf unserem Bioleiter-R-Paket namens CEMiTool (Russo et al., 2018) haben wir eine benutzerfreundliche webbasierte Anwendung entwickelt, die es Wissenschaftlern ohne Hintergrund in der Bioinformatik ermöglicht, umfassende Co-Expressions-Netzwerkanalysen durchzuführen.
Materialien und Methoden
Die Weboberfläche von webCEMiTool wurde entwickelt, um Benutzern die schnelle Erstellung umfassender Analysen zu ermöglichen, ohne dass ein bestimmtes Programm oder ein Internetbrowser installiert werden muss. Die einzige Voraussetzung für die Durchführung der modularen Analyse ist ein Datensatz, der die Expressionsniveaus aller Gene in Proben unter verschiedenen biologischen Bedingungen enthält (hierin definiert als “Klassen”). Es gibt keinen definierten Bereich Anzahl der Proben, aber unsere vorherige Studie schlägt ein Minimum von 15 Proben pro Datensatz vor (Russo et al., 2018). Obwohl es in erster Linie für Transkriptomdaten (d. H. RNA-seq oder Microarrays) entwickelt wurde, kann es möglicherweise auch zur Identifizierung von Modulen von Proteinen, Zytokinen und sogar Metaboliten verwendet werden. webCEMiTool wählt dann automatisch die Eingangsgene aus und identifiziert die Co-Expressionsmodule. Jedes Modul enthält eine Reihe von Genen, deren Expression einem ähnlichen Muster folgt.
Wir haben in webCEMiTool eine Funktion implementiert, die die Aktivität von Genmodulen für jede Probenklasse bewertet. Dazu müssen die Benutzer nur eine tabulatorgetrennte Textdatei für Beispielanmerkungen bereitstellen, die die Klasse jedes Beispiels informiert. Ein “Profildiagramm”, das den Medianwert der einzelnen Gene innerhalb des Moduls zeigt, wird dann im Abschnitt “Ergebnisse” des Tools angezeigt (Abbildung 1A).
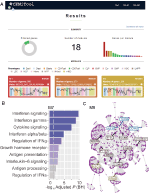
Abbildung 1. webCEMiTool Übersicht. (A) Zusammenfassung der webCEMiTool–Ergebnisse – Das Donut-Diagramm stellt den Anteil der ausgewählten Gene durch den unbeaufsichtigten Filter dar. Die Titelseite zeigt auch die Anzahl der erhaltenen Module sowie ein Balkendiagramm an, das die Anzahl der Gene in jedem Modul darstellt. Modulprofildiagramme veranschaulichen die mittlere Expressionsaktivität von Genen aus den Modulen in jeder Probe. Die Farben repräsentieren die verschiedenen Probenklassen. (B) Überrepräsentation Analyse – Dies zeigt die −log10 angepasst p-Wert (Benjamini-Hochberg) der angereicherten Pfade in einem Modul (Pfade definiert durch Benutzer eingegeben .gmt-Datei). (C) Gennetzwerk eines Moduls – Die am höchsten verbundenen Gene (Hubs) werden markiert und gefärbt, je nachdem, ob sie ursprünglich im Modul vorhanden waren (blau) oder aus einer vom Benutzer eingegebenen Interaktionsdatei eingefügt wurden (rot) oder beides (grün).
Um eine Funktionsanalyse zu ermöglichen, können die Benutzer auch überprüfen, ob die Genmodule mit bestimmten Signal- oder Stoffwechselwegen assoziiert sind (Abbildung 1B). Diese Pfade können leicht aus Datenbanken wie KEGG, Reactome und MySigDB extrahiert werden. Schließlich können Benutzer die Ergebnisse mit Interaktomdaten integrieren (d. H. Protein-Protein-Interaktionen, Transkriptionsfaktoren und ihre transkribierten Gene oder sogar miRNAs und ihre Zielgene). Mit dieser Funktion können Benutzer kritische Regulatoren von Modulen identifizieren (Abbildung 1C) und wertvolle Erkenntnisse für die experimentelle Validierung oder potenzielle Ziele für Arzneimittel gewinnen. Weitere Details zum Erhalt der optionalen Dateien finden Sie auf der Seite “Tutorial” der Website.2
Um zu demonstrieren, dass unsere Methode robust ist, führten wir eine beispiellose groß angelegte modulare Analyse mit über 1.000 öffentlich verfügbaren RNA-seq- und Microarray-Datensätzen und neuen RNA-seq-Daten von Patienten durch, die mit Leishmanien infiziert waren, unter Verwendung der CEMiTool R-Paketversion (Russo et al., 2018). Obwohl webCEMiTool und das Paket unterschiedliche Visualisierungsfunktionen haben und auf verschiedenen Plattformen basieren, ist die Kernfunktionalität von Co-Expression im Wesentlichen dieselbe. Das Online-Tool, das wir hier beschreiben, wurde entwickelt, um nicht programmierenden Forschern einen einfachen Zugang zu modularen Genanalysen zu ermöglichen, während die R-Bibliotheksversion auf Benutzer mit besseren Kenntnissen der Programmiersprache R ausgerichtet ist. Darüber hinaus besteht das Ergebnis-Dashboard aus interaktiven Diagrammen, die die Interpretation erleichtern. Darüber hinaus nutzt unser Tool das wachsende Ökosystem der Bioinformatik-Webdienste und stellt eine Schnittstelle zur Enrichr-Plattform her (Chen et al., 2013), so dass eine reichere Erfahrung für unsere Nutzer.
Ergebnisse
Wir haben gezeigt, dass webCEMiTool angewendet werden kann, um Expressionsdaten auf Einzelzellebene zu analysieren. Öffentlich zugängliche viscRNA-Seq-Daten (Virus-einschließlich Einzelzell-RNA-Seq) wurden aus der NCBI GEO-Datenbank (Zugangsnummer GSE110496) erhalten und als Eingabe für die Analyse verwendet. Die Daten beziehen sich auf das Transkriptom einzelner humaner Hepatomzellen (Huh7), die entweder mit dem Dengue-Virus (DENV) oder dem Zika-Virus (ZIKV) infiziert wurden, unter Verwendung von Multiplicity of Infection (MOI) 0, 1 oder 10 (Zanini et al., 2018). Zellen, die zu vier verschiedenen Zeitpunkten (4, 12, 24 und 48 h nach der Infektion) gesammelt wurden, wurden dann für die Einzelzell-Transkriptomanalyse mit einem angepassten Smart-seq2-Protokoll sortiert (Zanini et al., 2018). Der DENV-Datensatz umfasst 933 infizierte Zellen (MOI = 1 oder 10) und 303 Kontrollen (MOI = 0), während sich der ZIKV-Datensatz aus 488 infizierten Zellen (MOI = 1) und 403 Kontrollen zusammensetzt. Vor der Übermittlung der Analyse an die webCEMiTool-Plattform wurden beide Datensätze log10-transformiert und Gene, die nicht in mehr als 80% der Proben exprimiert wurden, entfernt. Die Datensätze wurden dann nach Virus und Zeitpunkt aufgeteilt und als Eingabe (Feld “Expression file”) in webCEMiTool verwendet. Zusätzlich zu den Genexpressionsdaten haben wir webCEMiTool auch die Probenphänotypen (d. H., Viruslasten) und Reaktom-Gensätze.
Unsere webCEMiTool-Analysen ergaben durchschnittlich sechs Module pro Zeitpunkt der DENV-Infektion und mehr als acht Module pro Zeitpunkt der ZIKV-Infektion. Repräsentativ für unsere Befunde haben wir pro Zeitpunkt ein Modul ausgewählt (Abbildung 2A). Es ist klar, dass 24 und 48 h nach der Infektion die Expressionsaktivität repräsentativer Module entsprechend der Viruslast zunimmt (Abbildung 2A). Als nächstes führten wir die Analyse der Pfadanreicherung der repräsentativen Module 24 h nach der Infektion mit dem webCEMiTool-Link für Enrichr durch (Abbildung 2B). Diese Ergebnisse bestätigen nicht nur das, was in der Originalpublikation (Zanini et al., 2018), liefern aber auch neue Erkenntnisse über die Physiopathologie von Dengue- und Zika-Virusinfektionen.
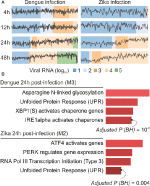
Abbildung 2. webCEMiTool angewendet auf Einzelzell-RNA-seq-Daten. (A) Profildiagramm von coexprimierten Genmodulen. Wir haben für jeden Zeitpunkt nach der Dengue-Virusinfektion (links) oder nach der Zika-Virusinfektion (rechts) ein repräsentatives Modul ausgewählt. Die schwarze Linie repräsentiert die mittlere Expressionsaktivität von Genen aus den Modulen über jede Probe. Die Farben repräsentieren die unterschiedliche Menge an Virus-RNA in der Zelle. (B) Überrepräsentation Analyse ausgewählter Module bei 24 h Post-Virus-Infektion. Die Balkendiagramme wurden aus dem mit webCEMiTool verknüpften Enrichr-Webtool angepasst. Die Balken sind proportional zum -log10 angepassten p-Wert (Benjamini-Hochberg) der angereicherten Pfade in einem Modul.
Diskussion
Obwohl nur wenige ähnliche webbasierte Anwendungen entwickelt wurden, um eine Koexpressionsgenanalyse durchzuführen (Tzfadia et al., 2016; Desai et al., 2017) liefern diese Tools keine vergleichbaren Ergebnisse wie webCEMiTool. Eine solche Anwendung ist GeNET (Desai et al., 2017). Dieses Webtool wurde entwickelt, um Gen-Co-Expressionsanalysen zu erleichtern und bietet Anreicherungsanalysen und Gen-zu-Gen-Netzwerke. Diese Analysen werden jedoch nur für drei Organismen (R. capsulatus, M. tuberculosis und O. sativa) durchgeführt. Ein weiteres Beispiel ist CoExpNetViz (Tzfadia et al., 2016), ein Webtool zur Visualisierung und Konstruktion von Gennetzwerken. Ähnlich wie GeNET ist CoExpNetViz in Bezug auf die Organismen etwas eingeschränkt, da es in erster Linie für pflanzliche Transkriptome entwickelt wurde. Das webCEMiTool zielt darauf ab, Co-Expressionsanalysen für jeden Organismus bereitzustellen. Obwohl CoExpNetViz als webbasierte Anwendung dargestellt wird, werden die Ergebnisse als komprimierter Ordner mit einer README-Datei an die Benutzer zurückgegeben.txt-Datei mit Anweisungen zur Visualisierung der Ergebnisse in der Cytoscape-App. Die Benutzer müssen dann die verschiedenen Ausgabedateien, die vom Tool bereitgestellt werden, manuell in Cytoscape einfügen. Diese zusätzlichen Schritte können den Prozess auch fehleranfällig und möglicherweise entmutigend für Benutzer machen, die mit Cytoscape nicht vertraut sind. Das webCEMiTool bietet wesentlich komfortablere browser-angezeigte Ergebnisse.
Wir haben auch gezeigt, dass webCEMitool in der Lage ist, Einzelzell-RNA-seq-Daten schneller und effizienter zu analysieren. Unsere Ergebnisse lieferten relevante Informationen über die biologischen Prozesse mit Dengue-und Zika-Virus-Infektion beteiligt. Alle diese Analysen wurden auf automatisierte und praktische Weise durchgeführt, ohne dass der Benutzer ein tiefes Verständnis für die interne Verarbeitung der Gen-Coexpressionsdatenanalyse haben muss.
Autorenbeiträge
LC, PR, BG-C und MA-P führten die Analysen durch. LC, GS-H und VM-C entwickelten das Webtool. HN konzipierte das Tool und überwachte die Arbeit. Alle Autoren helfen beim Schreiben des Papiers.
Interessenkonflikterklärung
Die Autoren erklären, dass die Forschung in Abwesenheit von kommerziellen oder finanziellen Beziehungen durchgeführt wurde, die als potenzieller Interessenkonflikt ausgelegt werden könnten.